



Les protéines sont essentielles à la vie, soutenant pratiquement toutes ses fonctions. Ce sont de grosses molécules complexes, constituées de chaînes d'acides aminés, et ce que fait une protéine dépend en grande partie de sa structure 3D unique (cadre 1). Déterminer dans quelles formes les protéines se replient est connu sous le nom de «problème de repliement des protéines» (cadre 2), et représente un défi majeur en biologie depuis 50 ans. Dans une avancée scientifique majeure, la dernière version de notre système d'IA AlphaFold (cadre 3) a été reconnue comme une solution à ce grand défi par les organisateurs de la biennale Critical Assessment of Protein Structure Prediction (CASP) [Évaluation critique de la prédiction de la structure des protéines]. Cette percée démontre l'impact que l'IA peut avoir sur la découverte scientifique et son potentiel pour accélérer considérablement les progrès dans certains des domaines les plus fondamentaux qui expliquent et façonnent notre monde.
La forme d'une protéine est étroitement liée à sa fonction, et la capacité de prédire cette structure permet de mieux comprendre ce qu'elle fait et comment elle fonctionne. Bon nombre des plus grands défis mondiaux, comme le développement de traitements contre les maladies ou la recherche d'enzymes qui décomposent les déchets industriels, sont fondamentalement liés aux protéines et au rôle qu'elles jouent.
PROFESSOR JOHN MOULT
CO-FONDATEUR ET PRÉSIDENT DE CASP, UNIVERSITÉ DU MARYLAND
Cela a été un centre de recherche scientifique intensive depuis de nombreuses années, en utilisant une variété de techniques expérimentales pour examiner et déterminer les structures de protéines, telles que la résonance magnétique nucléaire et la cristallographie aux rayons X. Ces techniques, ainsi que des méthodes plus récentes comme la cryo-microscopie électronique, dépendent d'essais et d'erreurs extensifs, qui peuvent prendre des années de travail minutieux et laborieux par structure, et nécessitent l'utilisation d'équipements spécialisés de plusieurs millions de dollars.
Le "problème du repliement des protéines''
Dans son discours de remerciement pour le prix Nobel de chimie 1972, Christian Anfinsen a postulé que, en théorie, la séquence primaire d'acides aminés d'une protéine devrait déterminer pleinement sa structure. Cette hypothèse a déclenché une quête de cinq décennies pour être en mesure de prédire par ordinateur la structure 3D d'une protéine en se basant uniquement sur sa séquence d'acides aminés 1D comme alternative complémentaire à ces méthodes expérimentales coûteuses et chronophages. Un défi majeur, cependant, est que le nombre de façons dont une protéine pourrait théoriquement se replier avant de s'installer dans sa structure 3D finale est astronomique. En 1969, Cyrus Levinthal a noté qu'il faudrait plus de temps que l'âge de l'univers connu pour énumérer toutes les configurations possibles d'une protéine typique par le calcul de la force brute - Levinthal a estimé à 10 ^ 300 conformations possibles pour une protéine typique. Pourtant, dans la nature, les protéines se replient spontanément, certaines en quelques millisecondes - une dichotomie parfois appelée paradoxe de Levinthal.
Explication du repliement des protéines
Résultats de l'évaluation CASP14
En 1994, le professeur John Moult et le professeur Krzysztof Fidelis ont fondé CASP en tant qu'évaluation à l'aveugle biennale pour catalyser la recherche, suivre les progrès et établir l'état de l'art dans la prédiction de la structure des protéines. C'est à la fois la référence en matière d'évaluation des techniques prédictives et une communauté mondiale unique fondée sur un effort partagé. Fondamentalement, le CASP choisit des structures de protéines qui n'ont été déterminées expérimentalement que très récemment (certaines étaient encore en attente de détermination au moment de l'évaluation) comme cibles pour les équipes afin de tester leurs méthodes de prédiction de structure; elles ne sont pas publiés à l'avance. Les participants doivent prédire aveuglément la structure des protéines, et ces prévisions sont ensuite comparées aux données expérimentales de vérité terrain lorsqu'elles deviennent disponibles. Nous sommes redevables aux organisateurs de CASP et à toute la communauté.
AlphaFold : la réalisation d'une percée scientifique
La principale métrique utilisée par CASP pour mesurer la précision des prévisions est le test de distance globale (GDT) qui va de 0 à 100. En termes simples, le GDT peut être approximativement considéré comme le pourcentage de résidus d'acides aminés (billes dans la chaîne protéique) à une distance seuil de la position correcte. Selon le professeur Moult, un score d'environ 90 GDT est considéré de manière informelle comme compétitif avec les résultats obtenus à partir de méthodes expérimentales.
Dans les résultats de la 14ème évaluation CASP, publiés aujourd'hui, notre dernier système AlphaFold atteint un score médian de 92,4 GDT global pour toutes les cibles. Cela signifie que nos prédictions ont une erreur moyenne (RMSD) d'environ 1,6 angströms, ce qui est comparable à la largeur d'un atome (ou 0,1 nanomètre). Même pour les cibles protéiques les plus dures, celles de la catégorie de modélisation libre la plus difficile, AlphaFold atteint un score médian de 87,0 GDT (données disponibles ici).

AMÉLIORATIONS DE LA PRÉCISION MÉDIANE DES PRÉDICTIONS DANS LA CATÉGORIE DE MODÉLISATION LIBRE POUR LA MEILLEURE ÉQUIPE DE CHAQUE CASP, MESURÉE COMME LE MEILLEUR DES 5 GDT.
DEUX EXEMPLES DE CIBLES PROTÉIQUES DANS LA CATÉGORIE MODÉLISATION LIBRE. ALPHAFOLD PRÉDIT DES STRUCTURES TRÈS PRÉCISES MESURÉES PAR RAPPORT AUX RÉSULTATS EXPÉRIMENTAUX.
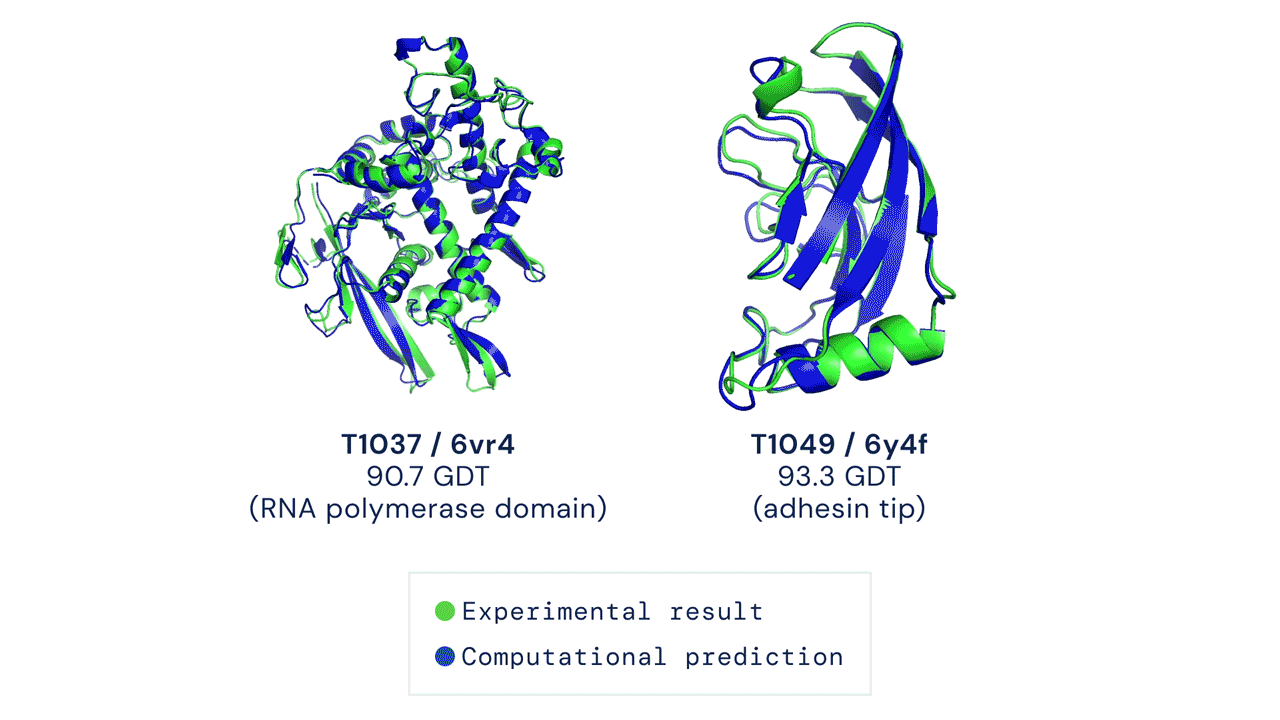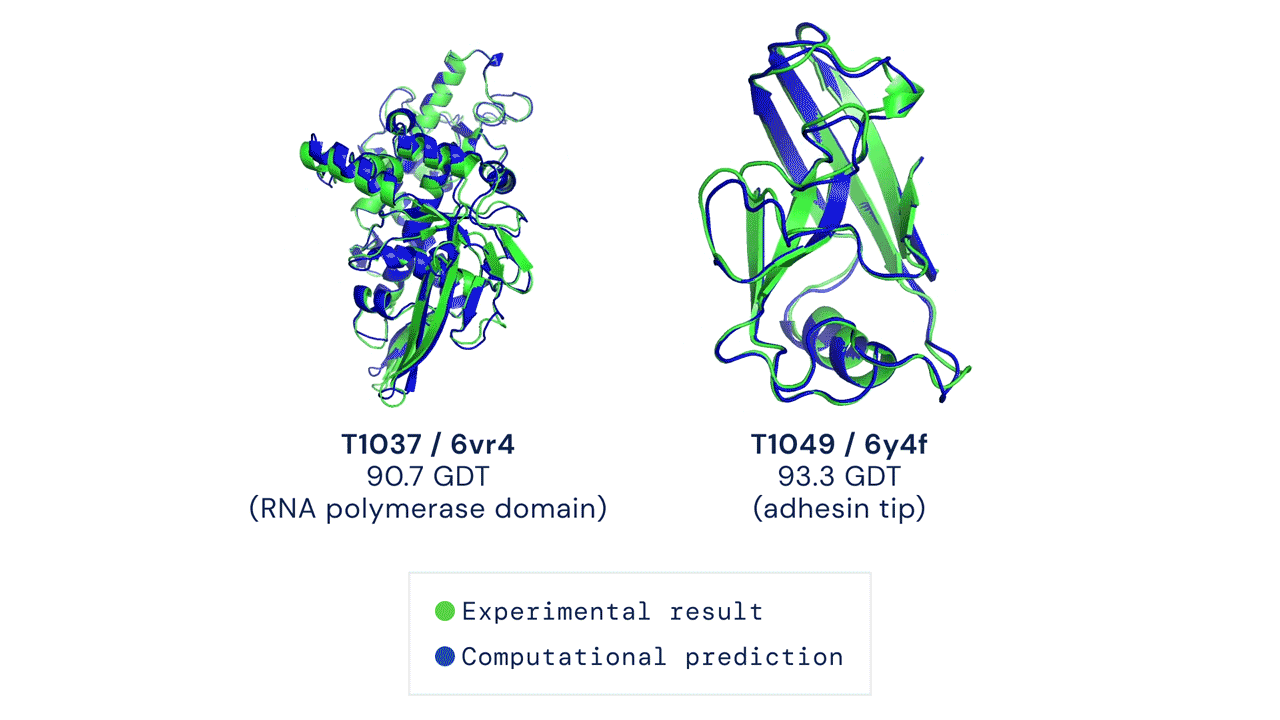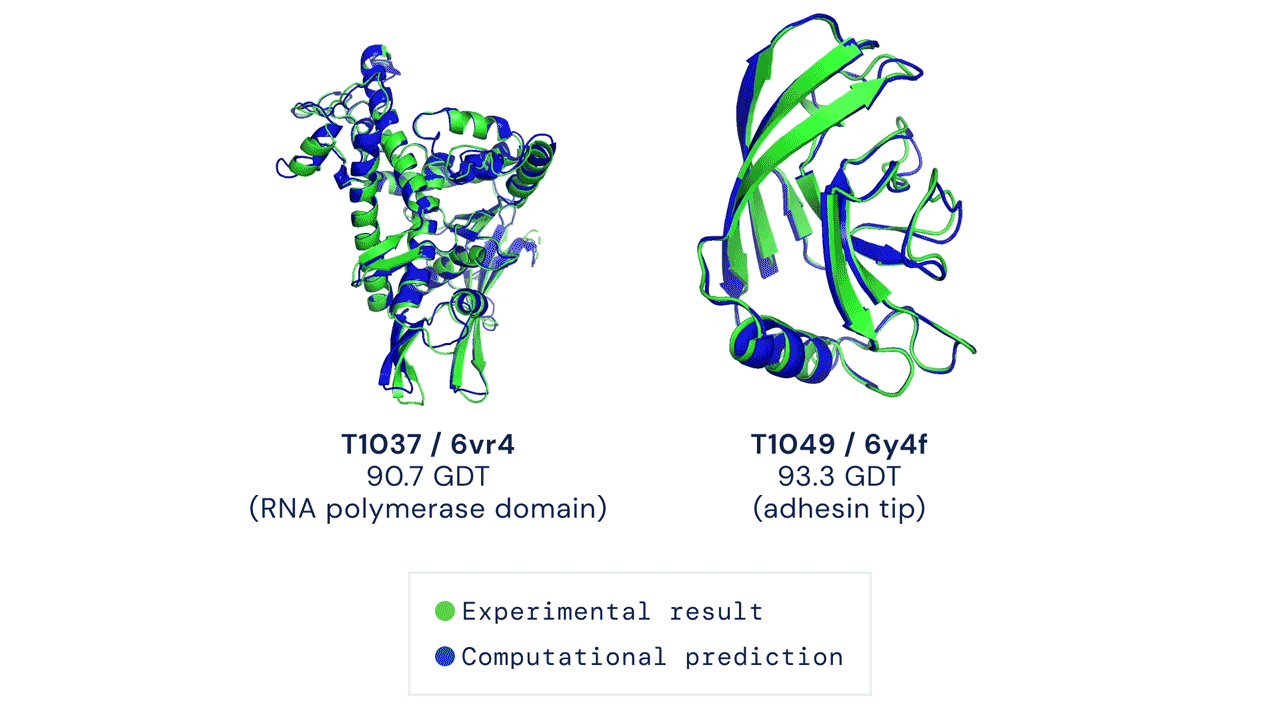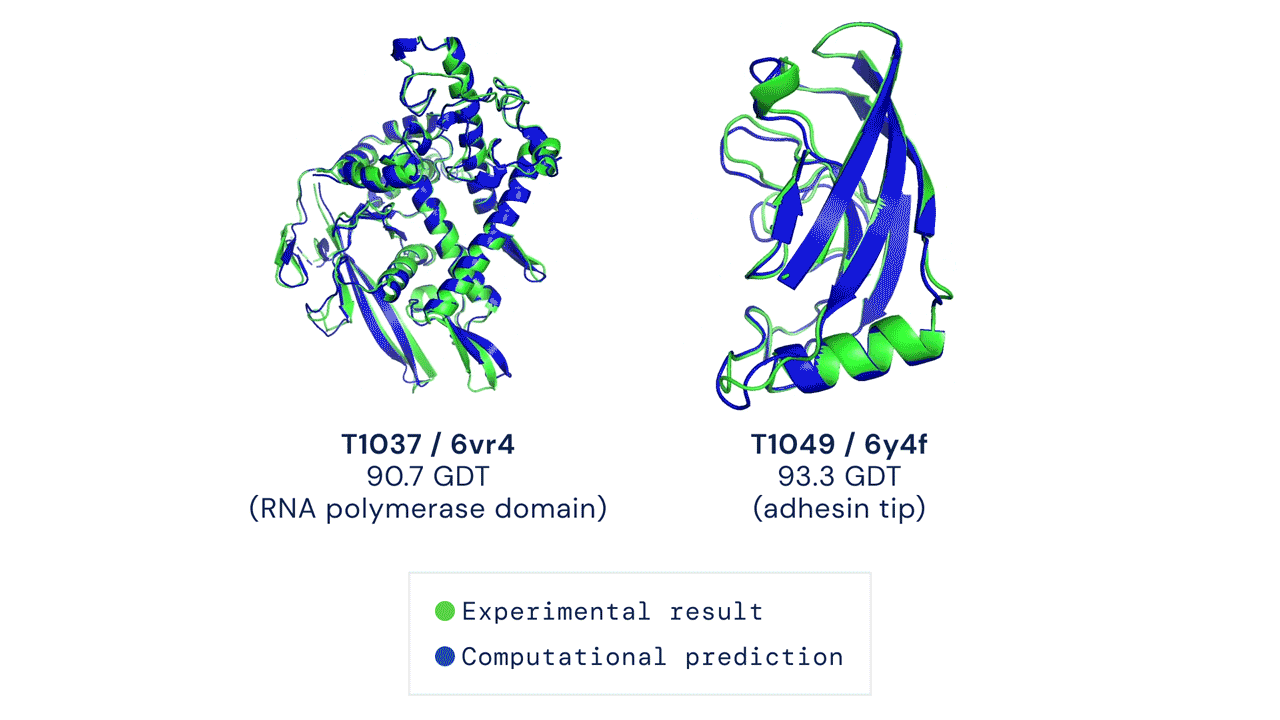
Ces résultats passionnants ouvrent le potentiel aux biologistes d'utiliser la prédiction de structure par ordinateur comme outil de base dans la recherche scientifique. Nos méthodes peuvent s'avérer particulièrement utiles pour des classes importantes de protéines, telles que les protéines membranaires, qui sont très difficiles à cristalliser et donc difficiles à déterminer expérimentalement.
PROFESSEUR VENKI RAMAKRISHNAN
LAURÉAT DU PRIX NOBEL ET PRÉSIDENT DE LA ROYAL SOCIETY
Notre approche du problème du repliement des protéines
Nous avons commencé CASP13 en 2018 avec notre version initiale d'AlphaFold, qui a atteint la plus grande précision parmi les participants. Ensuite, nous avons publié un article sur nos méthodes CASP13 dans Nature avec code associé, qui a inspiré d'autres travaux et implémentations open source développées par la communauté. Désormais, les nouvelles architectures d'apprentissage en profondeur que nous avons développées ont entraîné des changements dans nos méthodes pour CASP14, nous permettant d'atteindre des niveaux de précision inégalés. Ces méthodes s'inspirent des domaines de la biologie, de la physique et de l'apprentissage automatique, ainsi que, bien sûr, des travaux de nombreux scientifiques dans le domaine du repliement des protéines au cours du dernier demi-siècle.
Une protéine repliée peut être considérée comme un «graphe spatial», où les résidus sont les nœuds et les bords relient les résidus à proximité. Ce graphique est important pour comprendre les interactions physiques au sein des protéines, ainsi que leur histoire évolutive. Pour la dernière version d'AlphaFold, utilisée par CASP14, nous avons créé un système de réseau neuronal basé sur l'attention, formé de bout en bout, qui tente d'interpréter la structure de ce graphe, tout en raisonnant sur le graphe implicite qu'il construit. Il utilise des séquences liées évolutivement, un alignement de séquences multiples (MSA) et une représentation des paires de résidus d'acides aminés pour affiner ce graphique.
En répétant ce processus, le système développe de solides prédictions de la structure physique sous-jacente de la protéine et est capable de déterminer des structures très précises en quelques jours. De plus, AlphaFold peut prédire quelles parties de chaque structure protéique prédite sont fiables à l'aide d'une mesure de confiance interne.
Nous avons formé ce système sur des données accessibles au public constituées d'environ 170 000 structures protéiques de la banque de données protéiques ainsi que de grandes bases de données contenant des séquences protéiques de structure inconnue. Il utilise environ 128 cœurs TPUv3 (à peu près équivalents à ~ 100-200 GPU) exécutés sur quelques semaines, ce qui représente une quantité de calcul relativement modeste dans le contexte de la plupart des grands modèles de pointe utilisés dans l'apprentissage automatique aujourd'hui. Comme pour notre système CASP13 AlphaFold, nous préparons un article sur notre système à soumettre à une revue à comité de lecture en temps voulu.

UN APERÇU DE L'ARCHITECTURE PRINCIPALE DU MODÈLE DE RÉSEAU NEURONAL. LE MODÈLE FONCTIONNE SUR DES SÉQUENCES DE PROTÉINES LIÉES DE MANIÈRE ÉVOLUTIVE AINSI QUE SUR DES PAIRES DE RÉSIDUS D'ACIDES AMINÉS, PASSANT DE MANIÈRE ITÉRATIVE DES INFORMATIONS ENTRE LES DEUX REPRÉSENTATIONS POUR GÉNÉRER UNE STRUCTURE.
Le potentiel d'impact réel
Lorsque DeepMind a commencé il y a dix ans, nous espérions qu'un jour les percées de l'IA nous aideraient à servir de plate-forme pour faire progresser notre compréhension des problèmes scientifiques fondamentaux. Maintenant, après 4 ans d'efforts pour construire AlphaFold, nous commençons à voir cette vision se concrétiser, avec des implications dans des domaines comme la conception de médicaments et la durabilité environnementale.
Le professeur Andrei Lupas, directeur de l'Institut Max Planck pour la biologie du développement et évaluateur de CASP, nous a fait savoir que « les modèles étonnamment précis d'AlphaFold nous ont permis de résoudre une structure protéique sur laquelle nous étions bloqués pendant près d'une décennie, relançant nos efforts pour comprendre comment les signaux sont transmis à travers les membranes cellulaires. ».
Nous sommes optimistes quant à l'impact qu'AlphaFold peut avoir sur la recherche biologique et le monde en général, et nous sommes ravis de collaborer avec d'autres pour en savoir plus sur son potentiel dans les années à venir. En plus de travailler sur un article évalué par des pairs, nous explorons la meilleure façon de fournir un accès plus large au système de manière évolutive.
En attendant, nous examinons également comment les prévisions de la structure des protéines pourraient contribuer à notre compréhension de maladies spécifiques avec un petit nombre de groupes de spécialistes, par exemple en aidant à identifier les protéines qui ont mal fonctionné et à raisonner sur leur interaction. Ces informations pourraient permettre un travail plus précis sur le développement de médicaments, en complément des méthodes expérimentales existantes pour trouver plus rapidement des traitements prometteurs.
ARTHUR D. LEVINSON PHD,
FONDATEUR ET PDG CALICO, ANCIEN PRÉSIDENT-DIRECTEUR GÉNÉRAL, GENENTECH
Nous avons également vu des signes que la prévision de la structure des protéines pourrait être utile dans les efforts futurs de réponse à une pandémie, en tant que l'un des nombreux outils développés par la communauté scientifique. Plus tôt cette année, nous avons prédit plusieurs structures protéiques du virus SARS-CoV-2, dont ORF3a, dont les structures étaient auparavant inconnues. À CASP14, nous avons prédit la structure d'une autre protéine de coronavirus, ORF8. Un travail extrêmement rapide des expérimentateurs a maintenant confirmé les structures de l'ORF3a et de l'ORF8. Malgré leur nature difficile et ayant très peu de séquences associées, nous avons atteint un degré élevé de précision sur nos deux prédictions par rapport à leurs structures déterminées expérimentalement.
En plus d'accélérer la compréhension des maladies connues, nous sommes enthousiasmés par le potentiel de ces techniques pour explorer les centaines de millions de protéines pour lesquelles nous ne disposons pas actuellement de modèles - un vaste terrain de biologie inconnue. Puisque l'ADN spécifie les séquences d'acides aminés qui composent les structures protéiques, la révolution génomique a permis de lire des séquences protéiques du monde naturel à grande échelle - avec 180 millions de séquences protéiques et comptées dans la base de données Universal Protein (UniProt). En revanche, compte tenu des travaux expérimentaux nécessaires pour passer d'une séquence à une autre, seules 170 000 structures protéiques environ se trouvent dans la Protein Data Bank (PDB). Parmi les protéines indéterminées, il y en a peut-être avec des fonctions nouvelles et passionnantes et - tout comme un télescope nous aide à voir plus profondément dans l'univers inconnu - des techniques comme AlphaFold peuvent nous aider à les trouver.
Débloquer de nouvelles possibilités
AlphaFold est l'une de nos avancées les plus significatives à ce jour, mais, comme pour toute recherche scientifique, il reste encore à répondre à de nombreuses questions. Toutes les structures que nous prévoyons ne seront pas parfaites. Il reste encore beaucoup à apprendre, y compris comment plusieurs protéines forment des complexes, comment elles interagissent avec l'ADN, l'ARN ou de petites molécules, et comment nous pouvons déterminer l'emplacement précis de toutes les chaînes latérales d'acides aminés. En collaboration avec d'autres, il y a aussi beaucoup à apprendre sur la meilleure façon d'utiliser ces découvertes scientifiques dans le développement de nouveaux médicaments, des moyens de gérer l'environnement, etc.
Pour nous tous qui travaillons sur des méthodes informatiques et d'apprentissage automatique en science, des systèmes comme AlphaFold démontrent le potentiel étonnant de l'IA en tant qu'outil d'aide à la découverte fondamentale. Tout comme il y a 50 ans, Anfinsen a lancé un défi bien au-delà de la portée de la science à l'époque, de nombreux aspects de notre univers restent inconnus. Les progrès annoncés aujourd'hui nous donnent une confiance supplémentaire dans le fait que l'IA deviendra l'un des outils les plus utiles de l'humanité pour repousser les frontières de la connaissance scientifique, et nous attendons avec impatience les nombreuses années de travail acharné et de découvertes à venir !
Jusqu'à ce que nous ayons publié un article sur ce travail, veuillez citer :
Prédiction de la structure des protéines de haute précision à l'aide de l'apprentissage en profondeur
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Kathryn Tunyasuvunakool, Olaf Ronneberger, Russ Bates, Augustin Žídek, Alex Bridgland, Clemens Meyer, Simon AA Kohl, Anna Potapenko, Andrew J Ballard, Andrew Cowie, Bernardino Romera- Paredes, Stanislav Nikolov, Rishub Jain, Jonas Adler, Trevor Back, Stig Petersen, David Reiman, Martin Steinegger, Michalina Pacholska, David Silver, Oriol Vinyals, Andrew W Senior, Koray Kavukcuoglu, Pushmeet Kohli, Demis Hassabis.
Nous sommes au début de l'exploration de la meilleure façon de permettre à d'autres groupes d'utiliser nos prédictions de structure, tout en préparant un article évalué par des pairs pour publication. Bien que notre équipe ne soit pas en mesure de répondre à toutes les demandes, si AlphaFold peut être pertinent pour votre travail, veuillez envoyer quelques lignes à ce sujet à
Qu'est ce qu'une protéine ?
<haut de page>
Repliement des protéines
Le repliement des protéines est le processus physique par lequel une chaîne protéique acquiert sa structure tridimensionnelle native, une conformation qui est généralement biologiquement fonctionnelle, d'une manière rapide et reproductible. C'est le processus physique par lequel un polypeptide se replie dans sa structure tridimensionnelle caractéristique et fonctionnelle à partir d'une bobine aléatoire. Chaque protéine existe sous la forme d'un polypeptide déplié ou d'une bobine aléatoire lorsqu'elle est traduite d'une séquence d' ARNm en une chaîne linéaire d'acides aminés. Ce polypeptide est dépourvu de toute structure tridimensionnelle stable (durable) (le côté gauche de la première figure).
Lorsque la chaîne polypeptidique est synthétisée par un ribosome , la chaîne linéaire commence à se replier dans sa structure tridimensionnelle. Le pliage commence à se produire même pendant la traduction de la chaîne polypeptidique. Les acides aminés interagissent les uns avec les autres pour produire une structure tridimensionnelle bien définie, la protéine repliée (le côté droit de la figure), connue sous le nom d' état natif . La structure tridimensionnelle résultante est déterminée par la séquence d'acides aminés ou la structure primaire ( dogme d'Anfinsen ).
La structure tridimensionnelle correcte est essentielle pour fonctionner, bien que certaines parties des protéines fonctionnelles puissent rester dépliées, de sorte que la dynamique des protéines est importante. Le fait de ne pas se replier dans la structure native produit généralement des protéines inactives, mais dans certains cas, les protéines mal repliées ont une fonctionnalité modifiée ou toxique. Plusieurs maladies neurodégénératives et d' autres maladies sont censées résulter de l'accumulation de amyloïdes fibrilles formées par des protéines mal repliées. De nombreuses allergies sont causées par un repliement incorrect de certaines protéines, car le système immunitaire ne produit pas d' anticorps pour certaines structures protéiques.
La dénaturation des protéines est un processus de transition de l'état plié à l' état déplié. Cela se produit en cuisine, lors de brûlures, en protéinopathies et dans d'autres contextes.
La durée du processus de pliage varie considérablement en fonction de l'intérêt de la protéine. Lorsqu'elles sont étudiées à l'extérieur de la cellule, les protéines de repliement les plus lentes nécessitent plusieurs minutes ou heures pour se replier, principalement en raison de l'isomérisation de la proline, et doivent passer par un certain nombre d'états intermédiaires, comme des points de contrôle, avant que le processus ne soit terminé. D'un autre côté, de très petites protéines à domaine unique avec des longueurs allant jusqu'à cent acides aminés se replient généralement en une seule étape. Les échelles de temps en millisecondes sont la norme et les réactions de repliement des protéines connues les plus rapides sont terminées en quelques microsecondes.
<haut de page>

AlphaFold
AlphaFold peut prédire avec précision des modèles 3D de structures protéiques et a le potentiel d'accélérer la recherche dans tous les domaines de la biologie
Éléments de base de la vie
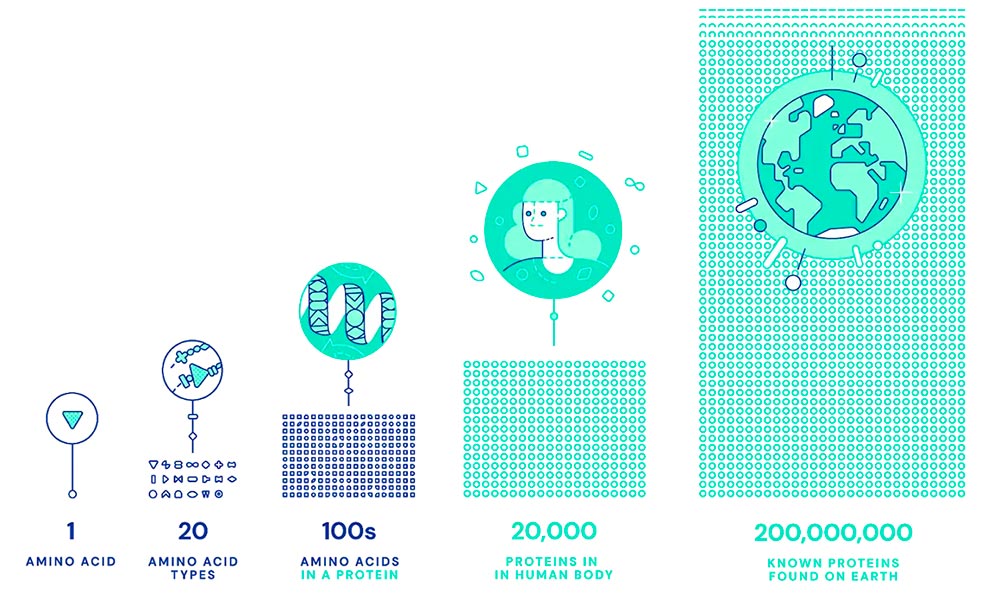
À l'intérieur de chaque cellule de votre corps, des milliards de minuscules machines moléculaires travaillent dur. C'est ce qui permet à vos yeux de détecter la lumière, à vos neurones de se déclencher et aux «instructions» de votre ADN à lire, qui font de vous la personne unique que vous êtes.
Ces machines exquises et complexes sont des protéines . Ils sous-tendent non seulement les processus biologiques de votre corps, mais tous les processus biologiques de chaque être vivant. Ce sont les éléments constitutifs de la vie.
Actuellement, il existe environ 200 millions de protéines connues , et 30 millions supplémentaires sont trouvées chaque année. Chacun a une forme 3D unique qui détermine son fonctionnement et son action.
Mais déterminer la structure exacte d'une protéine reste un processus coûteux et souvent long, ce qui signifie que nous ne connaissons que la structure 3D exacte d'une infime fraction des protéines connues de la science.
Trouver un moyen de combler cette lacune en expansion rapide et de prédire la structure de millions de protéines inconnues pourrait non seulement nous aider à lutter contre la maladie et à trouver plus rapidement de nouveaux médicaments, mais peut-être aussi à percer les mystères de la vie elle-même.
Explication du repliement des protéines
Le problème du repliement des protéines
Si vous pouviez démêler une protéine, vous verriez que c'est comme une chaîne de perles faite d'une séquence de différents produits chimiques appelés acides aminés.
Ces séquences sont assemblées selon les instructions génétiques de l' ADN d'un organisme.
L'attraction et la répulsion entre les 20 différents types d'acides aminés provoquent le repli de la corde dans un exploit d '« origami spontané », formant des boucles et plis complexes de la structure 3D d'une protéine.
Depuis des décennies, les scientifiques tentent de trouver une méthode pour déterminer de manière fiable la structure d'une protéine uniquement à partir de sa séquence d'acides aminés.
Ce grand défi scientifique est connu sous le nom de problème de repliement des protéines.
Qu'est AlphaFold ?
Nous avons commencé à travailler sur ce défi en 2016 et avons depuis créé un système d'IA appelé AlphaFold.
Il a été enseigné en lui montrant les séquences et les structures d'environ 100 000 protéines connues.
Notre dernière version peut maintenant faire des prédictions précises de la forme qu'une protéine formera en fonction de sa séquence d'acides aminés.
Il s'agit d'une avancée majeure et met en évidence l'impact que l'IA peut avoir sur la science.
Rejoindre une communauté de recherche mondiale
En 1994, des scientifiques intéressés par le repliement des protéines ont formé CASP (Critical Assessment of Protein Structure Prediction = Évaluation critique de la prédiction de la structure des protéines).
CASP est un forum communautaire qui permet aux chercheurs de partager les progrès sur le problème du repliement des protéines. La communauté organise également un défi biennal pour les groupes de recherche afin de tester l'exactitude de leurs prédictions par rapport à des données expérimentales réelles.
Les équipes reçoivent une sélection de séquences d'acides aminés pour des protéines dont la forme exacte en 3D a été mappée mais qui n'ont pas encore été publiées dans le domaine public. Les groupes doivent soumettre leurs meilleures prévisions pour voir à quel point ils sont proches des structures révélées ultérieurement.
Parmi les équipes qui ont participé à CASP13 (2018), AlphaFold s'est classé premier dans le défi de prédiction de la structure des protéines. Au CASP14 (2020), nous avons présenté notre dernière version d' AlphaFold, qui a maintenant atteint un niveau de précision considérable pour résoudre le problème de prédiction de la structure des protéines.
Notre travail s'appuie sur des décennies de recherche par les organisateurs de CASP et la communauté du repliement des protéines, et nous sommes redevables au nombre incalculable de personnes qui ont contribué aux structures protéiques au fil des ans, rendant possibles des évaluations aussi rigoureuses.
AlphaFold : la réalisation d'une percée scientifique
Comprendre Covid-19
Lorsque la Covid-19 est apparue, on en savait très peu à ce sujet. Mais des scientifiques du monde entier se sont réunis pour trouver des moyens de s'y attaquer.
Le SRAS-CoV-2, le virus qui cause la Covid-19, est composé d'environ 30 types de protéines, et une dizaine d'entre elles étaient mal comprises.
Notre équipe de recherche a utilisé AlphaFold pour prédire les structures de six protéines sous-étudiées dans le génome du virus SRAS-CoV-2, dans l'espoir qu'elles pourraient faire progresser notre compréhension du virus.
La structure de l'une de ces protéines , connue sous le nom d' ORF3a , a ensuite été élaborée à l'aide d'expériences scientifiques. Et dans le cadre de CASP14, nous avons démontré des prédictions encore plus précises pour ORF8 , une autre protéine SARS-CoV-2.
Ces résultats offrent un aperçu de la façon dont des outils d'IA comme AlphaFold pourraient mieux nous préparer à une future pandémie.
Accélérer la découverte scientifique
Un système comme AlphaFold capable de prédire avec précision la structure des protéines pourrait accélérer les progrès dans de nombreux domaines de recherche importants pour la société.
Par exemple, les informations limitées sur les structures des protéines ont été un obstacle majeur à l'amélioration de notre compréhension des maladies tropicales négligées comme la maladie du sommeil (trypanosomiase) et la leishmaniose , qui ont un impact sur la vie de millions de personnes et causent des dizaines de milliers de décès chaque année.
Elle freine également de nombreux efforts de recherche fondamentale. Par exemple, le développement d'un nouveau médicament peut prendre plus de 2,5 milliards de dollars et plus de 10 ans. AlphaFold pourrait contribuer à une découverte de médicaments meilleure et plus efficace en identifiant la structure de nombreuses protéines humaines impliquées dans la maladie.
Cela pourrait également aider à ouvrir de nouvelles possibilités telles que la recherche de protéines et d' enzymes qui décomposent les déchets industriels et plastiques ou capturent efficacement le carbone de l'atmosphère.
Il y a encore beaucoup de travail à faire avant de pouvoir contribuer à avoir un impact réel dans ces domaines et plus encore, mais le potentiel est énorme.
Si AlphaFold peut être pertinent pour votre travail, veuillez envoyer quelques lignes à ce sujet àCette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser. . Bien que notre équipe ne soit pas en mesure de répondre à chaque demande, nous serons en contact dans les cas où il y a place pour une exploration plus approfondie.
Regard vers le futur
Nos recherches sur AlphaFold se poursuivent, mais nos travaux jusqu'à présent - et les évaluations indépendantes d'organisations comme CASP - renforcent notre espoir que ses prédictions aideront bientôt à ouvrir de nouvelles possibilités dans la recherche biologique qui profiteront à la société.
Nous sommes enthousiasmés par cette prochaine phase du voyage d'AlphaFold et avons hâte de poursuivre notre travail avec la communauté scientifique mondiale pour libérer le potentiel des éléments constitutifs de la vie.
<haut de page>
traduction de l'article de DeepMind : https://deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology