


En collaboration avec IBM, nous (ForliLab) rejoignons le World Community Grid pour ancrer des millions de petites molécules à différentes protéines qui jouent un rôle dans le cycle de vie du SRAS-CoV2. Des molécules prometteuses seront testées expérimentalement par nos collaborateurs de Scripps et évolueront, espérons-le, pour devenir des antiviraux COVID-19.
Pour cela, nous allons cribler les liants conventionnels ainsi que les inhibiteurs covalents (c'est à dire irréversibles). En utilisant des réactions chimiques simples, de nombreuses molécules disponibles dans le commerce peuvent être modifiées pour porter un groupe réactif. Nous avons identifié des dizaines de millions de ces molécules dans la base de données ZINC. Maintenant, nous utilisons notre protocole Reactive Docking (voir encadré) pour trouver des molécules qui peuvent se fixer de manière covalente à la protéine. Les nouvelles molécules actives fourniront non seulement de nouveaux outils pour lutter contre l'infection, mais également des sondes moléculaires pour approfondir notre compréhension des nombreux aspects obscurs et encore inconnus de la biologie virale.
Grâce à l'infrastructure du World Community Grid et aux bénévoles qui font don de la puissance de calcul de leurs postes de travail, ordinateurs portables, téléphones portables et autres appareils, nous accosterons un très grand nombre de molécules, probablement le plus grand criblage covalent jamais réalisé.
Pour plus d'informations sur les laboratoires collaborateurs, les actualités, la FAQ et les statistiques, consultez la page OpenPandemics: COVID-19 Project (sur le site WCG) ou cette page du portail de L'Alliance Francophone.
La COVID-NineTeam (l'équipe COVID-19)
L'équipe est divisée en deux sous-équipes : l'équipe structure et l'équipe informatique.
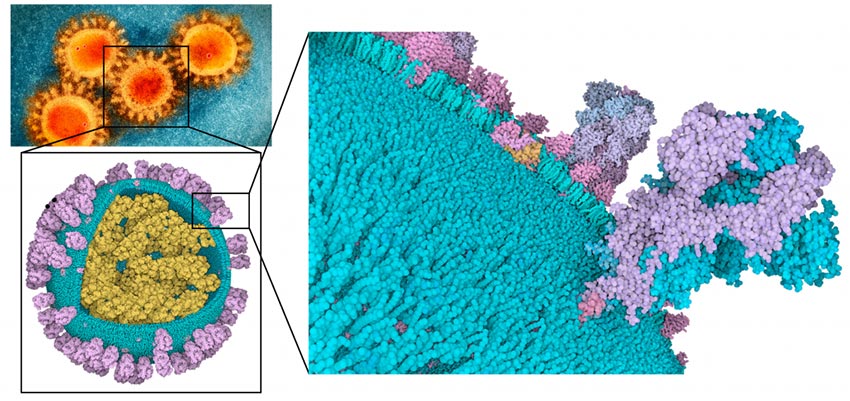
 L'équipe Structure organise et valide les structures protéiques cibles utilisées pour les amarrages. Ils analysent et préparent les structures à la recherche de poches de liaison potentielles ainsi que de résidus susceptibles d'être ciblés par les inhibiteurs covalents. Leur rôle est de garantir la qualité des structures cibles et de capter leur variabilité conformationnelle afin de garantir le succès des amarrages. L'équipe Structure travaille en collaboration avec Martina Maritan du Goodsell Lab pour construire des modèles à moyenne échelle du virus complet. Les images complètes du modèle viral ont été construites par Ludovic Autin.
L'équipe Structure organise et valide les structures protéiques cibles utilisées pour les amarrages. Ils analysent et préparent les structures à la recherche de poches de liaison potentielles ainsi que de résidus susceptibles d'être ciblés par les inhibiteurs covalents. Leur rôle est de garantir la qualité des structures cibles et de capter leur variabilité conformationnelle afin de garantir le succès des amarrages. L'équipe Structure travaille en collaboration avec Martina Maritan du Goodsell Lab pour construire des modèles à moyenne échelle du virus complet. Les images complètes du modèle viral ont été construites par Ludovic Autin.
 L'équipe informatique est responsable de la préparation des ligands à ancrer, de la préparation de la structure des composés disponibles dans le commerce et de la réalisation des énumérations in silico qui génèrent de nouvelles bibliothèques de dérivés synthétiquement accessibles, élargissant ainsi l'échantillonnage de l'espace chimique. Ils gèrent également l'infrastructure matérielle et logicielle pour gérer les données d'entrée et de sortie, effectuer les étapes d'analyse automatisées des résultats et les stocker dans la base de données.
L'équipe informatique est responsable de la préparation des ligands à ancrer, de la préparation de la structure des composés disponibles dans le commerce et de la réalisation des énumérations in silico qui génèrent de nouvelles bibliothèques de dérivés synthétiquement accessibles, élargissant ainsi l'échantillonnage de l'espace chimique. Ils gèrent également l'infrastructure matérielle et logicielle pour gérer les données d'entrée et de sortie, effectuer les étapes d'analyse automatisées des résultats et les stocker dans la base de données.
Pour plus de détails sur les personnes impliquées, consultez la page Membres du laboratoire.
Statistiques du projet OpenPandemics
- traduction de la page du site ForliLab : https://forlilab.org/openpandemics-covid-19/?linkId=89465168
Recherche d'intérêts
L'objectif de nos recherches est d'utiliser des outils informatiques pour démêler et comprendre les règles physico-chimiques des processus biologiques.
Nous développons de nouvelles méthodes de calcul de pointe qui nous permettent de prédire et d'analyser les interactions de petites molécules organiques (synthétiques ou naturelles) avec des structures macromoléculaires biologiques telles que les protéines et l'ADN / ARN.Nous appliquons ces outils en combinaison avec d'autres techniques de modélisation moléculaire pour étudier et interpréter la nature des événements biologiques d'un point de vue mécanistique et thermodynamique, et explorer rapidement l'espace chimique pour identifier les modulateurs moléculaires des cibles thérapeutiquement pertinentes. Nous avons un certain nombre de collaborations au sein de Scripps Research ainsi qu'avec des chercheurs d'autres institutions académiques et des industries pharmaceutiques.
Suite AutoDock
Nous développons des outils informatiques pour la conception de médicaments basés sur la structure, en particulier un logiciel d'ancrage moléculaire. AutoDock et AutoDock Vina sont des logiciels open-source et font partie de la suite la plus utilisée, précédemment développée dans le Molecular Graphics Lab, maintenant CCSB . Nous développons la prochaine génération de moteurs d'ancrage et les outils de dépistage virtuels, concevons des potentiels énergétiques améliorés, de meilleures estimations de l'énergie libre de liaison et de nouveaux protocoles pour la conception de médicaments.
L'objectif est de rendre l'ancrage plus précis et utile en le rendant plus rapide et plus significatif chimiquement. Nous améliorons la description des termes chimiques, physiques et thermodynamiques pour inclure les préférences de torsion des liaisons rotatives, une description plus précise des interactions non covalentes et, plus important encore, des effets de solvatation. Pour cela, nous nous tournons vers les champs de force modernes, les méthodes de structure électronique et les simulations de dynamique moléculaire. Dans le même temps, en conjonction avec le calcul GPU, nous explorons de nouveaux algorithmes de recherche pour naviguer dans le paysage énergétique.
Publications pertinentes
Cartographie de l'anisotropie des liaisons hydrogène (JCTC, 2020)
Amarrage computationnel protéine-ligand et criblage virtuel de médicaments avec la suite AutoDock (Nat.Protoc.2016)
Un champ de force avec des eaux discrètes déplaçables et une entropie de désolvatation pour l'amarrage de ligands hydratés (JMedChem 2012)
AutoDock4Zn: un champ de force AutoDock amélioré pour l'amarrage de petites molécules aux métalloprotéines de zinc (JChemInfMod 2014)
AutoDock Bias: amélioration de la prédiction du mode de liaison et du criblage virtuel en utilisant les interactions connues protéine-ligand (Bioinformatics, 2019)
Accélération d'AutoDock4 avec les GPU et la recherche locale basée sur les dégradés (2019)
D3R Grand Challenge 4: prédiction de pose prospective des ligands BACE1 avec AutoDock-GPU (2019)
Les partenaires
Le développement de notre code est effectué en collaboration avec des partenaires de l'industrie du matériel et des logiciels.
Ancrage à haut débit d'inhibiteur covalent
La modélisation des liants covalents n'est pas triviale, en particulier d'une manière à haut débit. Plusieurs méthodes covalentes sont maintenant disponibles, mais la majorité de ces approches sont efficaces lorsque l'information sur les structures cibles est connue (c'est-à-dire le site de liaison covalente).
Poussés par la nécessité de surmonter cette barrière, nous concevons la méthode Reactive Docking, une approche qui permet la prédiction prospective des sites de liaison covalente sur une protéine, à travers la simulation de l'événement de réaction entre le résidu covalent et l'ogive du ligand. De plus, Reactive Docking permet de modéliser différents résidus et différents types d'ogives.
Cette méthode a été appliquée avec succès à plusieurs cibles et elle convient au HTVS des modificateurs covalents pour la découverte de nouveaux sites de liaison covalente sur différentes protéines.
Publications pertinentes
Découverte de ligands covalents à l'échelle du protéome dans des systèmes biologiques natifs (Nature, 2016)
Stratégie de «découverte de médicaments inversés» pour identifier les protéines ciblées par les électrophiles latents, comme en témoignent les fluorosulfates d'aryle (JACS, 2018)
Amarrage covalent à l'aide de l'autodock: méthodes d'attracteur à deux points et de chaîne latérale flexible (ProtSci 2018)
Découverte agnostique activée par SuFEx d'inhibiteurs covalents de l'élastase des neutrophiles humains (PNAS 2019)
Profilage global de la réactivité et de la ligandabilité de la lysine dans le protéome humain (NatChem 2017)
Structure intégrative des rayons X et modélisation moléculaire pour la rationalisation de la puissance et de la sélectivité des inhibiteurs de la procaspase-8 (ACS ChemBio 2020)
Cartographie accélérée du protéome ligandable à l'aide de paires de sondes énantiomères entièrement fonctionnalisées (NatChem 2019)
Cycle de vie du VIH-1
La recherche de médicaments pouvant interférer avec différentes étapes du cycle de vie du VIH-1 reste un sujet de grand intérêt, car les médicaments actuellement disponibles ne sont pas en mesure d'éradiquer l'infection. Nous ciblons la protéine Capside du VIH-1 en mettant en œuvre une stratégie basée sur l'ancrage conventionnel et réactif pour le criblage virtuel de petites molécules qui pourraient moduler l'assemblage viral. L'approche d'amarrage réactif est utilisée pour cribler de petites molécules avec une ogive SuFEx, qui pourraient se lier étroitement à la poche identifiée.
Le laboratoire Forli fait partie du HIV Interaction in Viral Evolution Center (HIVE).
Publications pertinentes
Base structurelle pour la liaison de l'inhibiteur de transfert de brin aux intasomes du VIH (Science, 2020)
Modélisation intégrative du complexe de ribonucléoprotéines VIH-1 (PLoS Comput.Bio.2019)
Une nouvelle classe d'inhibiteurs de l'intégrase du VIH-1 allostérique identifiés par criblage de fragments cristallographiques du domaine du noyau catalytique (JBC 2016)
Distinguer les liants des faux positifs par des calculs d'énergie gratuits: dépistage des fragments contre le site du volet de la protéase du VIH (J.Phys.Chem.B.2015)
Prédiction aveugle de la liaison de l'intégrase du VIH à partir du défi SAMPL4 (JCAMD 2014)
Découverte cristallisée de médicaments à base de fragments cristallins: utilisation d'une bibliothèque de fragments bromés ciblant la protéase du VIH (Chem.Biol.Drug Des, 2014)
Modèles moléculaires 3D de virions VIH-1 entiers générés avec cellPACK (Faraday Discuss.2014)
Une nouvelle interaction intersubunit critique pour l'assemblage de base du VIH-1 définit une poche de liaison d'inhibiteur potentiellement ciblable (mBio 2019)
Applications de modélisation moléculaire
Application de techniques de modélisation moléculaire, comme la dynamique moléculaire, les pharmacophores moléculaires et les criblages virtuels.
Publications pertinentes
Base structurelle de la puissance et de l'efficacité modifiées affichées par un major in vivo du métabolite de la pioglitazone, un médicament PPARγ antidiabétique (J.Med.Chem 2019)
Interaction humanisée du facteur GPIbα – von Willebrand chez la souris (Blood Adv.2018)
L'acide anacardique produit naturel des noix de cajou stimule la production de pièges extracellulaires de neutrophiles et l'activité bactéricide (JBC, 2016)
La Phosphorylation Directionnelle Et Le Transport Nucléaire Du Facteur D'épissage SRSF1 Est Régulé Par Un Motif De Reconnaissance D'ARN (JBC, 2016)
…
- traduction de la page ForliLab : https://forlilab.org/research/