


Caractérisation d'états partiellement ordonnés dans le domaine N-terminal intrinsèquement désordonné de p53 à l'aide de simulations de dynamique moléculaire à la milliseconde
La dernière publication scientifique obtenue avec votre soutien et vos calculs est ici :
Herrera-Nieto, P., Pérez, A. & De Fabritiis, G.Caractérisation d'états partiellement ordonnés dans le domaine N-terminal intrinsèquement désordonné de p53 à l'aide de simulations de dynamique moléculaire millisecondes . Sci Rep 10, 12402 (2020).
Il étudie un problème ouvert et difficile en biologie, à savoir la relation structure-fonction des protéines intrinsèquement désordonnées (IDP). Les protéines IDP sont impliquées dans de nombreux processus de régulation, et pourtant sont difficiles à étudier à la fois expérimentalement et par ordinateur. La quantité d'énergie fournie par les volontaires de GPUGRID est venue à la rescousse, permettant un échantillonnage très étendu de p53, un IDP exemplaire impliqué dans des processus oncogènes.
- article lié -
Caractérisation d'états partiellement ordonnés dans le domaine N-terminal intrinsèquement désordonné de p53 en utilisant des simulations de dynamique moléculaire à la milliseconde
Résumé
L'exploration des protéines intrinsèquement désordonnées en isolement est une étape cruciale pour comprendre leur comportement dynamique complexe. En particulier, l'émergence d'états partiellement ordonnés n'a pas été explorée en profondeur. La caractérisation expérimentale de ces états partiellement ordonnés reste insaisissable en raison de leur nature transitoire. La dynamique moléculaire atténue cette limitation grâce à sa capacité à explorer des échelles de temps biologiquement pertinentes tout en conservant une résolution atomique. Ici, des simulations de dynamique moléculaire non biaisées à la milliseconde ont été réalisées dans la région N-terminale exemplaire de p53. En combinaison avec des modèles d'état de Markov de pointe, les simulations ont révélé l'existence de plusieurs états partiellement ordonnés représentant ∼ 40% de la population à l'équilibre. Certains des états les plus pertinents présentent des conformations hélicoïdales similaires à la structure liée de p53 à Mdm2, ainsi que de nouveaux éléments de feuille-β. Cela met en évidence la complexité potentielle qui sous-tend la surface énergétique des protéines intrinsèquement désordonnées.
Introduction
Au cours des dernières décennies, la compréhension de la fonction des protéines a été résumée par le triumvirat séquence-structure-fonction : les séquences de protéines codent des plis capables d'effectuer des tâches spécifiques. Les protéines intrinsèquement désordonnées (IDP) défient ce principe en assurant la médiation de leurs fonctions biologiques malgré l'absence d'une structure tridimensionnelle stable. Un tel comportement configure une surface énergétique relativement plane où coexistent de nombreuses conformations isoénergétiques. Cette surface peut être modifiée dans une certaine mesure, comme le révèle le déplacement vers certaines sous-populations observé dans la formation de complexes protéine-IDP ou molécule-IDP. De même, les paramètres cinétiques régissant les conversions entre les sous-populations peuvent également être modifiés par des modifications post-traductionnelles. Ainsi, la surface énergétique des IDP est loin d'être constituée exclusivement de conformations enroulées au hasard, et des éléments de preuve soutiennent l'existence d'états partiellement ordonnés. La caractérisation de ces états partiellement ordonnés est cruciale pour comprendre la fonction des IDP, leurs mécanismes d'action et leur modulation potentielle.
L'hétérogénéité structurelle des PDI se résume à une collection ou un ensemble de conformations. Elles peuvent être résolues expérimentalement en utilisant la résonance magnétique nucléaire (RMN) ou des données de diffusion des rayons X aux petits angles. La principale limite des ensembles de PID résolus de cette manière est qu'ils se concentrent sur des moyennes globales plutôt que de plonger dans des coordonnées atomiques particulières. Il existe également de nombreuses approximations de calcul pour résoudre cette tâche. Elles impliquent généralement une étape initiale de génération de conformateurs, suivie d'une étape de raffinement qui minimise les différences entre la bibliothèque générée et les données expérimentales. Cependant, de nombreux ensembles résolus par calcul peuvent correspondre aux mêmes observations expérimentales.
Les simulations de dynamique moléculaire (MD) ont été largement utilisées au fil des ans pour naviguer sur des surfaces énergétiques complexes dans d'autres problèmes biologiques, c'est-à-dire dans le pliage, la liaison protéine-protéine et la modulation des IDP par des modifications post-traductionnelles ou par l'interaction avec leurs partenaires pliés. Dans le contexte des ensembles IDP, les simulations MD ont été principalement appliquées comme outil de génération conformationnelle. Néanmoins, l'objectif principal de la MD dans ce domaine serait de définir des ensembles fiables sans qu'il soit nécessaire de recourir à des procédures de biais ou de repondération. Dans cette optique, des études récentes ont utilisé des méthodes d'échantillonnage améliorées, telles que le MD Hamiltonian replica exchange, pour définir des ensembles IDP correspondant aux informations expérimentales disponibles. En termes de temps agrégé, l'étude s'est déroulée pour ∼ 10 μs, tandis que d'autres ont réalisé des simulations plus poussées, ∼ 200 μs mais dans une seule trajectoire.
Les technologies actuelles permettent aux simulations de MD d'atteindre des temps agrégés de l'ordre de la milliseconde , ce qui fait de cet outil un outil précieux pour l'exploration des systèmes biologiques à des échelles de temps de plus en plus longues. Le potentiel offert par les simulations de MD à haut débit couplées à l'analyse des modèles d'état de Markov (MSM) pour l'exploration des paysages conformationnels des PDI a été testé dans certains peptides sujets à l'agrégation. Le principal avantage offert par ce tandem est la possibilité d'aborder des sous-populations au sein d'ensembles et d'étudier la cinétique qui les contrôle, plutôt que de travailler avec des moyennes de population. En se concentrant sur les sous-populations les plus pertinentes et sur leurs propriétés cinétiques, il est possible de se faire une idée de l'émergence d'états partiellement ordonnés dans les détails atomistiques.
Nous utilisons ici des simulations de MD complètes et non biaisées et des MSM de pointe pour explorer la variabilité structurelle de la région N-terminale de p53 de manière isolée. p53 est une protéine largement étudiée, étant donné sa relation avec les processus oncogènes. Elle comprend des sections désordonnées aux deux extrémités N et C, qui interagissent avec divers partenaires. La courte région comprenant les résidus 10 à 40 est particulièrement importante. Elle forme une hélice α stable lors de son interaction avec la protéine Mdm et a été le sujet principal de nombreuses études informatiques et expérimentales. Le complexe p53-Mdm2 a servi de modèle pour le développement de médicaments peptidomimétiques et de référence privilégiée pour plusieurs études de médecine visant à reconstruire le processus de liaison et la cinétique associée. Des études de RMN et de SAXS de la région N-terminale isolée ont révélé un profil d'hélicité similaire à celui observé dans le complexe p53-Mdm2, ce qui implique que les conformations liées pourraient également être échantillonnées avant la liaison.
Les principaux résultats montrent l'existence de nombreux états cinétiquement pertinents, représentant ∼ 40% de la population à l'équilibre, y compris des niveaux élevés d'éléments structurels secondaires. En particulier, les simulations montrent la présence d'une hélice α-états enrichis similaire à la pose pliée trouvée dans le complexe avec Mdm2, ainsi que, une interaction enchevêtrée entre la formation de β-strands menant à de nouvelles structures enrichies de β-sheet. Dans l'ensemble, cela illustre la complexité des états partiellement ordonnés dans l'espace conformationnel d'un IDP exemplaire, tel que la région N-terminale de p53.
Résultats et débats
Identification des états enrichis de la structure secondaire
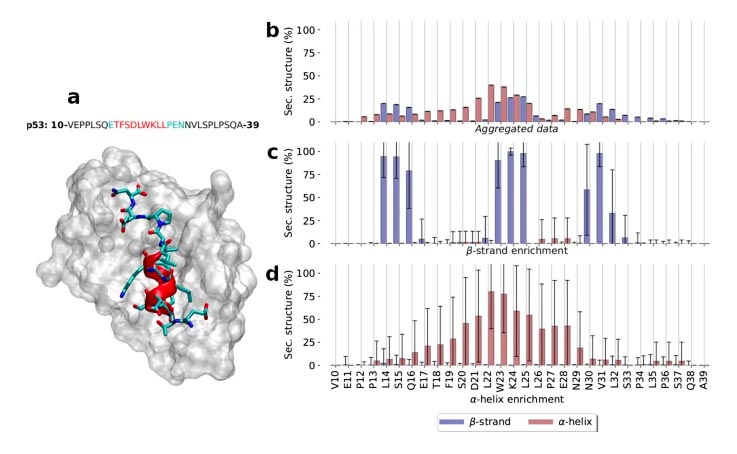
Le temps de simulation de l'exécution du MD s'est élevé à ∼ 1,4 ms. Dans un premier temps, la structure secondaire du MD agrégé a été analysée. Les données ont montré la coexistence des deux brins α-helix et β-strand, chacun atteignant un pic à ∼20 % dans la région centrale de la protéine (Fig. 1b). Le profil d'hélicité suit une distribution en forme de cloche, tandis que le brin β est plus faiblement dispersé en trois groupes à proximité des résidus S15, K24 et V31.
Les données de MD ont été utilisées pour créer une MSM basée sur la matrice d'autodistance backboneCα+sidechainO,N qui divise l'espace en 11 ensembles différents de conformations cinétiquement liées, appelées macroétats (étiquetés M1-11). Les sous-populations de MSM séparent avec succès des ensembles métastables de conformations enrichies dans chaque type de structure secondaire (Fig. 1c,d), ce qui implique que ces éléments structurels apparaissent de manière concertée, plutôt que d'être la moyenne des propensions structurelles indépendantes des résidus.
Le profil d'hélicité affiché par l'état enrichi en hélice correspond à la conformation liée de p53 lors de l'interaction avec Mdm2 (Fig. 1a). Il passe du résidu T18 à L26, et les niveaux maximums d'hélicité se trouvent dans W23-un acide aminé essentiel pour cette interaction. Des profils similaires ressortent des études de RMN30. Outre cet état, beaucoup d'autres présentent également divers degrés d'hélicité (Fig. S1). La tendance des PID à acquérir des profils de structure secondaire ressemblant à leur conformation repliée a également été observée dans d'autres PID, et elle a également été liée au mécanisme de liaison à leur partenaire et à leurs propriétés de signalisation.
Pour le brin β, la séparation des éléments structurels secondaires en leurs propres états devient particulièrement évidente dans M2, où trois brins β - à savoir β1, β2 et β3 du terminal N au terminal C - sont organisés en une double feuille antiparallèle (Fig. 1c), définissant l'état partiellement ordonné avec le niveau de structure le plus significatif. Outre les macro-états enrichis de structure secondaire mentionnés ci-dessus, de nombreux autres états présentent également des profils différents de β-brin et α-hélix (Fig. S1 et S2). Il s'agit notamment d'un certain nombre d'États présentant différentes dispositions de la feuille β, avec un seul brin, soit β1-β2 soit β2-β3. Dans l'ensemble, cela met en évidence la variété des configurations possibles dans le paysage conformationnel de la p53.
Figure 1
p53 secondary structure propensities (a) p53–MDM2 complex: MDM2 protein is shown as a white surface. p53 is depicted as cyan sticks and the helical region between residues 18 and 26 as red cartoon, (PDB code: 1YCR). Above, the sequence of p53 used for the simulation is displayed: in red the helical section, in cyan the rest of the peptide found in the PDB structure, and in black the extended sequence. Secondary structure profiles derived from MD data: β-strand and α-helix profiles for the (b) aggregated data and for those macrostates of the MSM enriched in either (c) β-sheet and (d) α-helix.
Caractérisation cinétique du paysage conformationnel de la p53
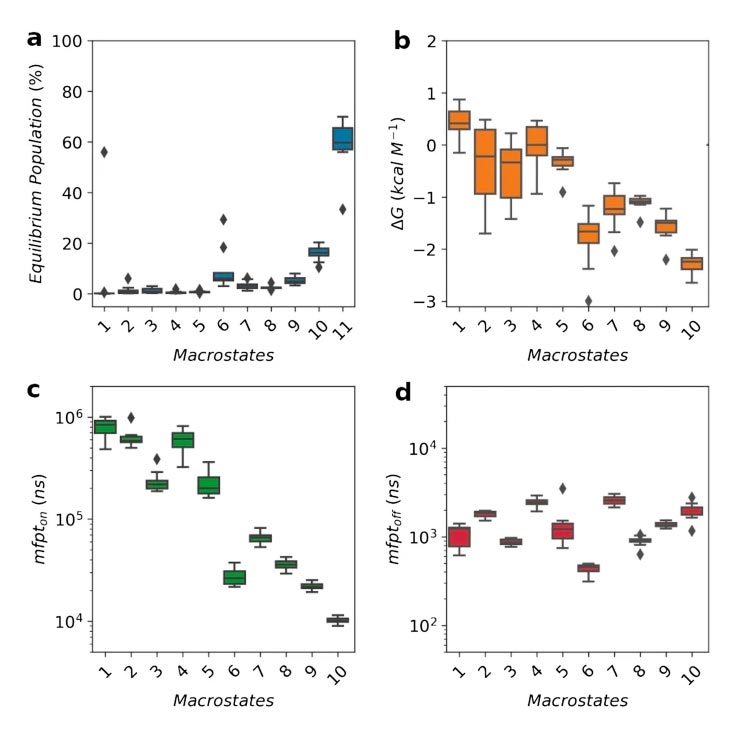
Les états partiellement ordonnés représentent une proportion importante de la population d'équilibre (∼ 40%, Fig. 2a). Les macroétats à triple brin, les plus repliés, ont une faible population (< 1%), contrairement aux états à double brin comme M10, qui atteignent ∼ 20% à l'équilibre. Cependant, l'état le plus peuplé - M11 ; avec ∼ 60 % de la population - est structurellement hétérogène, et n'a aucun élément structurel secondaire ou contacts à longue distance. Par conséquent, les états partiellement ordonnés ne sont pas énergétiquement favorisés par rapport aux configurations les plus étendues, et leurs énergies libres vont de 0,5 à - 2,5 k cal M-1 (Fig. 2b). Ceci peut être visualisé plus en détail dans la surface d'énergie de p53 (Fig. S3). Il y a deux minima bien définis séparés par une petite barrière énergétique. L'un d'eux est couvert par les états étendu et hélicoïdal (M6,9,11), et l'autre par les conformations β2-β3 (M10). Les zones de haute énergie sont occupées par les états les plus structurés, comme M2. Un tel profil, avec de nombreux états énergétiquement similaires, correspond à la description des personnes déplacées à l'intérieur de leur propre pays, prises isolément. Le niveau de compactage de p53 suit une tendance similaire (Fig. S4). Les états les plus effondrés ont un rayon de giration proche de celui attendu pour une protéine repliée. Les plus flexibles combinent des sections rigides et non rigides et échantillonnent des conformations étendues. L'absence de surcompactage de l'ensemble est conforme aux capacités des champs de force de pointe.
Figure 2
Estimation de (a) la population d'équilibre, (b) ΔG, (c) m f pton, (d) m f ptoff, pour chaque macrostate après 10 tours de bootstrap. Les paramètres cinétiques pour M11 (structurellement hétérogène) ne sont pas indiqués car il a été utilisé comme état source pour les calculs.
Les profils de population distincts observés précédemment ont un impact sur le comportement cinétique des macro-états. Il existe une différence d'environ deux ordres de grandeur entre les estimations de m f pton maximales et minimales, qui séparent les macroétats plus rapides/plus peuplés (M6-10) des macroétats plus lents/moins peuplés (M1-5) (Fig. 2c). Les macro-états plus rapides comprennent la conformation hélicoïdale et plusieurs états à double brin. Les états plus lents, en revanche, comprennent les états à trois brins et d'autres états peu peuplés. Les taux d'arrêt restent similaires dans tous les macro-états (Fig. 2d).
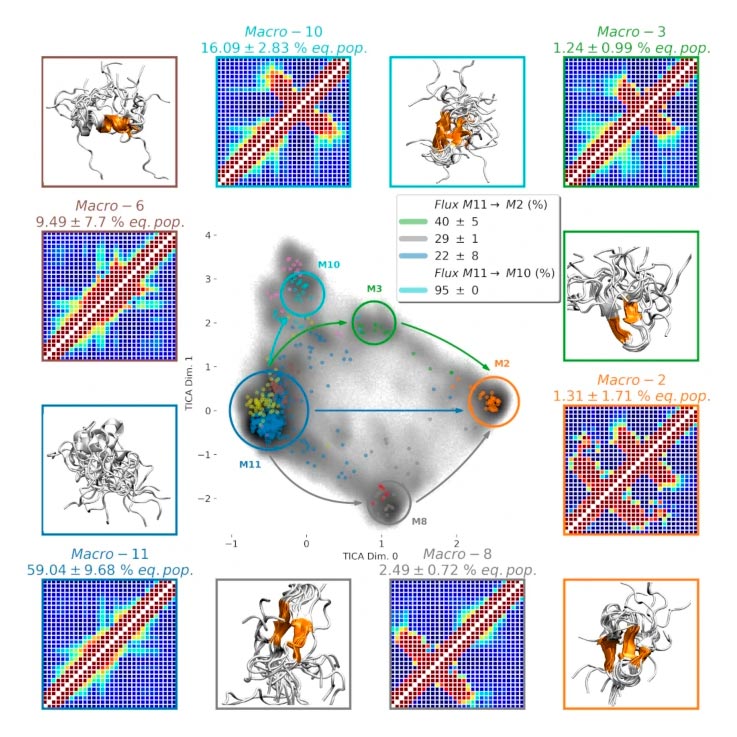
Nous avons utilisé la théorie des chemins de transition pour étudier les chemins et les flux les plus pertinents pour l'interconversion des macro-états. En particulier, l'objectif était d'élucider le processus de repliement menant de l'état moins structuré à la conformation triple brin. Trois voies principales (Fig. 3-panneau central) sont impliquées dans ce processus. Le chemin le moins transité représente ∼ 15% du flux total, et atteint directement la conformation pliée à partir de la conformation étendue. D'autre part, les chemins les plus transités impliquent la participation d'intermédiaires double-brin, la structure β1-β2 prenant ∼ 40% et la conformation β2-β3 étant responsable des 30% restants du flux. En outre, d'autres états enrichis de β, tels que la fiche étendue β2-β3 que l'on trouve dans M10, sont déconnectés de ce réseau et peuvent être directement atteints à partir de M11 sans avoir besoin d'intermédiaires. Il est intéressant de souligner que certaines conformations (telles que M3,10), malgré leur similarité structurelle, présentent un ralentissement ∼ 30 qui explique les différences de stabilité susmentionnées.
Figure 3
p53 paysage conformationnel. Le panneau central illustre les deux premières dimensions de l'espace TICA. En gris, un histogramme 2D 200×200 cases représente le nombre de trames des données MD agrégées. Les microétats MSM sont répartis en fonction de leur centre et colorés par rapport à leur macro-état correspondant. Les flèches représentent les principales voies menant du macro-état le plus étendu (Macro-11) aux macro-états les plus enrichis de la feuille β (Macro-2 et Macro-10). La correspondance entre l'emplacement des macrostate dans le panneau central et les panneaux latéraux est représentée par des couleurs. Les panneaux latéraux décrivent les macro-états en termes de cartes de contacts des résidus. La visualisation des protéines est réalisée en superposant 20 structures utilisant des résidus mis en évidence en orange pour l'alignement des structures.
En résumé, les états partiellement ordonnés qui peuplent le paysage conformationnel sont structurels et cinétiquement divers. Les états coexistent à des échelles de temps différentes, même s'ils sont structurellement similaires, comme c'est le cas de M6,11. Ces deux états présentent une feuille courte et une feuille étendue β2-β3 mais ont des valeurs de kon de 5⋅105 M-1 s-1 et 1⋅107 M-1 s-1 respectivement.
Comparaison avec les données RMN
Afin d'assurer et de valider les observations de MD, les données de simulation ont été comparées à des décalages chimiques (CS) de la colonne vertébrale déterminés expérimentalement pour la région N-terminale de p53. Les CS expérimentaux ont été résolus pour la longueur complète de l'extrémité N-terminale (résidus 1-93), mais seuls les CS des résidus 10 à 39 ont été utilisés (pour correspondre à la séquence simulée). CS permet d'inférer des tendances de structure secondaire des résidus sur des protéines pliées et désordonnées. Les calculs ont été effectués à l'aide de deux logiciels, SPARTA+ et SHIFTX2, sur un ensemble de 2 000 structures sélectionnées au hasard en fonction des probabilités d'équilibre des macrostats. Les CS calculées avec les deux programmes ont donné des résultats similaires. Dans l'ensemble, il existe une forte corrélation entre les valeurs expérimentales et les valeurs de CS calculées par MD pour Cα, Cβ, et N avec des valeurs R2 de 0,98, 0,99 et 0,88 (Fig. S5). Les différences entre les CS expérimentales et calculées restent dans les limites de l'erreur d'estimation intrinsèque de chaque logiciel (∼1 p.p.m pour SPARTA+ et 0,4, 0,5 et 1,1 pour Cα, Cβ et NH, respectivement, pour SHIFTX2), ce qui indique que les réarrangements structurels observés dans les données MD sont conformes à ceux déterminés par les expériences RMN.
Conclusion
La caractérisation du paysage conformationnel p53 à l'aide de simulations de MD non biaisées a révélé un nombre élevé d'états transitoires partiellement ordonnés représentant ∼ 40% des populations d'équilibre. L'ordre partiel résulte de la formation des éléments structurels de α-helix et β-strand. L'état hélicoïdal ressemble à la structure acquise par p53 lors de l'interaction avec Mdm2. Le MSM a également montré la présence de plusieurs états enrichis de β, non décrits précédemment, qui ont établi des contacts à long terme par l'arrangement d'une ou deux feuilles de β. Ces processus sont différents sur le plan cinétique, et certains des états les plus rapides sont rapidement accessibles à partir du macro-état enroulé au hasard et très peuplés à l'équilibre. Ainsi, il serait possible pour certains d'entre eux de jouer des rôles biologiquement pertinents et pourrait même fournir de nouvelles stratégies pour la modulation des PDI. D'autres études de calcul avec p53 ont également laissé entendre la présence de conformations effondrées dans cette région. L'exploration des PDI sujets à l'agrégation, tels que le bêta-amyloïde et le hIAPP, a montré la formation spontanée d'états métastables β-hairpin, mais avec de très petites populations.
L'étude actuelle fournit une description structurelle et cinétique détaillée du paysage conformationnel d'un PDI en utilisant des simulations de MD en combinaison avec des MSM. Étant donné le nombre élevé de motifs linéaires courts au sein du protéome humain, un pipeline similaire pourrait, en principe, être appliqué plus largement afin d'étudier si d'autres PDI peuvent également partager des comportements aussi complexes. Cependant, le fait d'atteindre un temps de simulation de quelques millisecondes pourrait ne pas être adapté dans des études plus approfondies avec des cibles multiples. De nouvelles techniques d'échantillonnage adaptatives, qui permettent une exploration plus intelligente des surfaces, pourraient atténuer ce problème en réduisant le temps de calcul nécessaire pour obtenir des résultats similaires. Quelques considérations finales sur la tâche sont l'effort de calcul pour simuler des peptides plus longs ainsi que la nécessité de comparer avec des données expérimentales pour valider les états partiellement repliés et leurs temps de relaxation. Nous utilisons ici une section relativement courte de 30 acides aminés de p53, alors que des domaines complètement désordonnés peuvent spammer des centaines de résidus.
Méthodes
Mise en place d'une simulation de la dynamique moléculaire
Afin de réaliser l'exploration de l'espace conformationnel de p53, de vastes simulations parallèles ont été effectuées. La région sélectionnée de p53 a été spammée des résidus 10 à 39.
Un ensemble de 110 structures a été utilisé comme conformations initiales pour la course MD (Fig. S8c). Tous les systèmes ont été construits avec VMD (version 1.9.2, http://www.ks.uiuc.edu/Research/vmd/), solvatés avec TIP3P (chaque système comprenait ∼ 8 200 molécules d'eau, ce qui donne une concentration finale de protéines de ∼ 6,8 mM), avec une concentration finale de NaCl de 0,05 M. Un intégrateur Langevin avec une constante d'amortissement de 0,1 ps-1 a été utilisé. L'étape d'intégration a été fixée à 4 fs, les atomes d'hydrogène lourds étant mis à l'échelle jusqu'à quatre fois leur masse naturelle. Les calculs électrostatiques ont été effectués à l'aide de PME avec une distance de coupure de 9 Å et un espacement de grille de 1 Å. L'équilibrage a été effectué à 300 K, en subissant tout d'abord 250 étapes de minimisation de l'énergie, suivies de simulations de 0,1 ns dans un ensemble NVE (la pression a été maintenue à 1 atm en utilisant le barostat berendesen) et de 2 ns dans un ensemble NPT. Nous avons utilisé le champ de force CHARMM22*, une modification du CHARMM22 original avec des potentiels de torsion de la colonne vertébrale ajustés pour produire des conformations plus étendues. Après équilibrage, aucun isomère de la proline cis n'a été détecté.
La production de 1 μs a été réalisée à 310 K à l'aide du projet de calcul distribué GPUGrid utilisant le moteur ACEMD (inclus dans le HTMD).
Analyse du modèle d'état de Markov
Les séries de production ont généré un total de 1 337 trajectoires (chaque système équilibré a été utilisé au moins 10 fois) de 1μs chacune, ce qui est similaire. Ainsi, les runs de production ont pris en compte un temps de simulation agrégé de ∼ 1,4 ms afin de maximiser l'exploration de l'espace conformationnel. Toutes les analyses de données de MD ont été effectuées à l'aide du HTMD (version 1.22.0 https://github.com/Acellera/htmd).
Les données du MD ont été utilisées pour construire un MSM. L'analyse a été réalisée en intégrant les coordonnées atomiques comme matrice d'autodistance entre Cα et les chaînes latérales des atomes d'azote et d'oxygène. Ensuite, la méthode d'analyse en composantes indépendantes du temps (TICA) a réduit la dimensionnalité des données à un temps de latence fixe de 20 trames. Les paramètres des dernières étapes de construction ont été sélectionnés sur la base des scores du quotient de Rayleigh de la matrice généralisée (GMRQ) (Fig. S6 et S7). Un nombre final de 9 dimensions TICA et 600 clusters ont été sélectionnés. Les grappes de données ont été définies à l'aide de l'algorithme MiniBatchKMeans.
Les micro-états ont été fusionnés avec un temps de retard de 120 ns, en suivant le tracé des échelles de temps implicites (Fig. S8a) en 11 macroétats (en utilisant l'algorithme PCCA+ et en se basant sur la discrétisation de l'espace TICA montré dans la Fig. S8e). Le test de Chapman-Kolmogrov (Fig. S9) a confirmé que les paramètres donnent un modèle markovien. Enfin, la théorie des chemins de transition a été utilisée pour calculer les flux entre les états.
Pour chaque mesure, l'erreur a été estimée en créant 10 répliques bootstrap indépendantes du MSM en utilisant un ensemble aléatoire contenant 80% des trajectoires.
Calculs du déplacement chimique
Les calculs des décalages chimiques dérivés des MD ont été effectués en utilisant 2 000 trames réparties entre les macro-états en fonction de leur probabilité d'équilibre. L'entrée 17760 de la banque de données sur la résonance magnétique biologique a été utilisée pour obtenir des données expérimentales sur les déplacements chimiques pour la région N-terminale de p53. L'expérience a été réalisée avec la pleine longueur du N-terminal (résidus 1 à 93), mais pour la comparaison, nous avons utilisé la section 10-39. Deux logiciels différents ont été utilisés : SPARTA+ (version 2.90, http://spin.niddk.nih.gov/bax/software/SPARTA+/index.html) et SHIFTX2 (version 1.09, http://www.shiftx2.ca/).
traduction du message du forum GPUGrid : https://www.gpugrid.net/forum_thread.php?id=5156
et de l'article publié (cadre gris) : https://www.nature.com/articles/s41598-020-69322-2