


Récapitulatif
Dans cette mise à jour, l'équipe Forli Lab de Scripps Research nous donne un aperçu des trois protéines virales qu'ils étudient actuellement et décrit leurs projets pour un avenir proche.
Contexte
Notre équipe de Scripps Research effectue des simulations de modélisation moléculaire pour rechercher des candidats potentiels pour le développement de traitements contre la COVID-19. Pour réussir, nous nous sommes associés à World Community Grid pour la puissance de calcul massive dont nous avons besoin pour réaliser des millions d'expériences simulées en laboratoire.
Dans cette mise à jour, nous vous donnerons un aperçu des trois protéines virales que nous étudions actuellement, nous exposerons nos plans pour étudier plus de protéines avec l'aide du World Community Grid et nous vous indiquerons où nous en sommes pour fournir des traitements potentiels à nos collaborateurs de laboratoire en vue des derniers tests. (Nous sommes proches !)
Mais d'abord, le membre de l'équipe de recherche, le Dr Martina Maritan, a créé une infographie sur le projet pour aider à décrire le processus de recherche complet.
Vous pouvez cliquer ici pour voir les détails complets.

De plus, voici les définitions de quelques termes pour vous aider à mieux comprendre notre mise à jour.
- Pose de ligand : Une pose de ligand est l'un des arrangements possibles pour une molécule donnée. Dans le cadre de ce projet, nous avons plusieurs poses de ligand pour chaque composé chimique, et nous choisissons la meilleure.
- Amarrage moléculaire : Ce processus est l'étude de la façon dont deux molécules ou plus s'assemblent, comme la façon dont une protéine dans une cellule humaine ou dans un virus s'accorde avec un composé chimique. Dans OpenPandemics - COVID-19, nous pouvons tirer parti de ce processus à grande échelle, grâce à la puissance de calcul massive de World Community Grid, en criblant virtuellement des millions de composés chimiques pour voir lesquels pourraient être capables de se lier aux protéines du virus SARS-Cov2 qui cause la COVID -19.
- Protéines virales : Ce sont des protéines générées par un virus (dans ce projet, par SARS-Cov2).
Gestion et analyse des données
Dans la mise à jour précédente, nous avons détaillé comment nous avons réussi à compresser les résultats plus que possible avec la compression de données traditionnelle en ne transmettant que les variables de chaque pose de ligand (que nous appelons techniquement "génome"). Ceci est une brève mise à jour sur ce qui se passe après que nous «réhydratons» ces poses, ou en d'autres termes, comment nous analysons les résultats.
Les données que vous créez tous sont analysées en plusieurs étapes. La première étape se produit lorsque nous recevons des packages (ou des lots d'unités de travail) de World Community Grid. Nous analysons chaque pose de ligand qu'ils contiennent (chaque paquet contient environ un demi-million de poses) et capturons les interactions spécifiques avec la protéine cible. Ces interactions peuvent être des liaisons hydrogène, des interactions non polaires ou, surtout pour l'amarrage réactif, la survenue de la réaction chimique entre les ogives et les résidus ciblés.
Ces informations, ainsi que le génome des résultats, sont ce que nous stockons dans notre base de données de résultats pour la prochaine étape de l'analyse. Actuellement, grâce à votre effort dédié, vous avez créé un total d'environ 6,8 milliards de poses de ligands occupant environ 20 To de stockage. Bien qu'il s'agisse de beaucoup de données - et nous avons récemment mis à jour notre serveur de base de données avec plus de stockage (250 To !) Pour cette raison même - c'est encore à peu près autant que les coordonnées individuelles pour chaque pose le demanderaient. Bien que cette étape de l'analyse soit la plus coûteuse en calcul, nous sommes actuellement en mesure d'analyser environ quatre fois plus de paquets par jour que ce que nous recevons en utilisant un seul nœud de calcul.
La deuxième étape d'analyse comprend plusieurs niveaux de filtrage sur l'ensemble des résultats, en utilisant les interactions que nous avons capturées dans la première phase. Nous partons d'un large filtre basé sur les énergies des ligands et les interactions avec les résidus d'intérêt pour une cible donnée. Cette requête de base de données renvoie des identificateurs de pose individuels. Ces poses sont ensuite classées en fonction des critères de sélection qu'elles remplissent. Ces critères de sélection sont spécifiques à la cible et incluent des éléments tels que « a des interactions avec x mais aucune avec y », « des liaisons hydrogène avec z », et d'autres. Enfin, les poses de ligand qui répondent à tous les critères les plus importants sont réhydratées (c'est-à-dire que leurs coordonnées 3D réelles sont restaurées) et stockées en tant que candidats potentiels. Actuellement, cette étape de l'analyse donne environ 20 000 candidats potentiels sur les 6,8 milliards de poses de ligand.
La dernière étape de l'analyse consiste à sélectionner manuellement les molécules les plus prometteuses pour des tests expérimentaux par nos collaborateurs. Le processus de sélection consiste en une inspection visuelle des poses ancrées pour évaluer quelques aspects nécessaires à la liaison de ces molécules à la protéine, mais qui souvent ne sont pas correctement modélisés par ancrage. La complémentarité de forme est l'un de ces aspects : la molécule doit s'intégrer parfaitement dans la poche protéique, sans espace vide excessif entre la molécule et la protéine. Un autre aspect important est la souche - la molécule ne doit pas avoir d'interaction défavorable avec elle-même pour tenir dans la poche.
Sur les 20 000 candidats potentiels, nous avons sélectionné environ 70 molécules pour des tests expérimentaux et nous allons les commander bientôt ! (Voir la section ci-dessous « Rendre les molécules virtuelles RÉELLES » . )
Paquets actuels
Ci-dessous : un rendu de la protéine nsp3
Ci-dessous : un rendu de la protéine nsp5
Ci-dessous : un rendu de la protéine nsp15
Les packages actuels ciblent trois protéines virales (voir les vidéos ci-dessus créées par le Dr Jérome Eberhardt, membre de l'équipe de recherche): nsp3, nsp5 et nsp15. C'est la première fois que des molécules sont ancrées contre nsp5 et nsp15.
Toutes les molécules contiennent le groupe acrylamide qui peut être capable de réagir avec certaines cystéines dans les protéines virales. Toutes ces molécules contenant de l'acrylamide sont disponibles dans la base de données ZINC ou directement auprès de nos fournisseurs de produits chimiques, garantissant ainsi leur prix raisonnable ou leur synthèse.
De nouvelles cibles
Alors que nous continuons à préparer des packages pour les cibles actuelles, nous en explorons déjà de nouvelles pour lesquelles nous organiserons les prochaines projections. L'une d'elles est la protéine non structurelle 8 (nsp8).
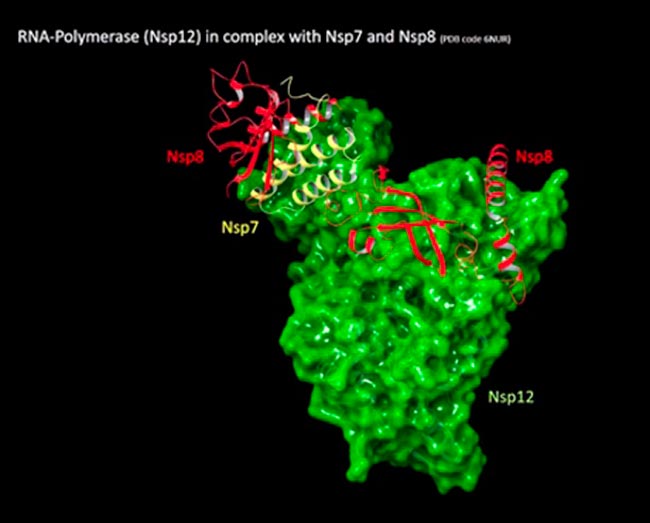
À l'instar d'autres coronavirus, le SARS-CoV-2 présente 16 protéines non structurales hautement conservées et présentant différentes fonctions, dont la formation du complexe de réplication-transcription. Parmi elles, Nsp8 est une protéine non structurale coronavirale qui, avec nsp7, interagit avec et régule l'ARN-polymérase ARN-dépendante (nsp12, ou RdRp), qui catalyse la synthèse de l'ARN viral pendant l'infection. Essentiellement, nsp8 et nsp7 agissent comme des cofacteurs stimulant l'activité polymérase de nsp12, jouant un rôle clé dans le cycle de réplication et de transcription du virus.
D'un point de vue structurel, nsp8 assume différentes conformations en fonction du partenaire en interaction, en particulier, il se trouve dans une «conformation proche» lorsqu'il est lié à nsp7 et dans une «conformation ouverte» lorsqu'il est lié à nsp12. Nous avons commencé notre enquête à partir du complexe ternaire nsp7-nsp8-nsp12 (Pdb id : 6NUR ) en se concentrant dans un premier temps uniquement sur la protéine nsp8 à l'interface avec nsp12, qui présente deux résidus de cystéine 114 et 142 respectivement.
En collaboration avec le Gervasio Lab, nous avons décidé d'étudier l'accessibilité de ces poches contenant les résidus de cystéine à l'aide de simulations dynamiques aqueuses et co-solvants. Par la suite, une sélection minutieuse des structures nsp8 dérivées de la dynamique sera ensuite utilisée comme cibles d'entrée pour le criblage virtuel des bibliothèques de fragments réactifs.
Rendre les molécules virtuelles RÉELLES
Nous travaillons avec le fournisseur de produits chimiques Enamine qui fournira les analogues physiques des molécules qui ont été ancrées à l'aide du World Community Grid. Ces molécules proviennent de leur base de données REAL, un ensemble de molécules pour lesquelles elles ont une voie de synthèse prévue, mais qui n'ont pas forcément été synthétisées dans le passé. Leur catalogue REAL contient 1,36 milliard de molécules que nous pouvons immédiatement cribler et synthétiser à la demande. Cela nous permet d'explorer davantage l'espace chimique que nous ne le pourrions avec les bibliothèques physiques existantes.
Cela signifie que nous enverrons bientôt des molécules à nos collaborateurs pour valider nos résultats de criblage et, espérons-le, nous rapprocher des premiers inhibiteurs !
Merci à tous ceux qui soutiennent OpenPandemics - COVID-19 !
Par : L'équipe de recherche OpenPandemics
29 oct. 2020
traduction de l'article WCG : https://www.worldcommunitygrid.org/about_us/viewNewsArticle.do?articleId=661&linkId=103200949