


Récapitulatif
AutoDock, le logiciel qui anime OpenPandemics et d'autres projets World Community Grid, a été créé il y a 30 ans chez Scripps Research.
Apprenez-en davantage sur ce logiciel puissant dans cet article et dans le document de recherche ci-joint récemment publié.
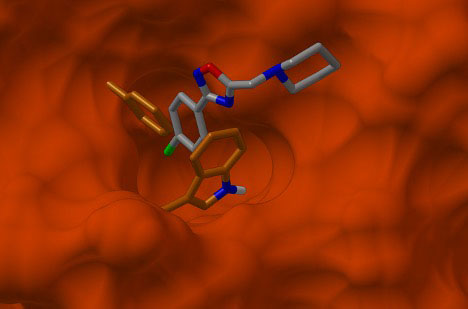
L'image ci-dessus, générée avec Python Molecular Viewer,
montre le résultat d'une simulation d'amarrage moléculaire effectuée avec AutoDock.
La recherche scientifique computationnelle était un domaine différent il y a quelques décennies, en grande partie à cause des limites de l'espace de stockage et de l'infrastructure informatique. Mais alors que la recherche de nouveaux et meilleurs traitements pour des maladies telles que le SIDA devenait plus urgente, les scientifiques de Scripps Research ont créé la première version d'AutoDock au début des années 1990.
Aujourd'hui, AutoDock a été utilisé dans de nombreuses études scientifiques dans des institutions et des entreprises pharmaceutiques du monde entier, notamment dans les projets FightAIDS@Home et OpenPandemics - COVID-19 du World Community Grid. Si ces projets sont tous deux axés sur la recherche de traitements contre le SIDA et COVID-19, respectivement, AutoDock a également été utilisé dans des études sur la méthodologie de conception des médicaments (ou la manière de concevoir des traitements).
AutoDock a été affiné et amélioré au cours des trois dernières décennies. Nombre de ces changements sont décrits dans un article sur l'histoire et l'évolution d'AutoDock publié dans la revue Protein Science. Vous pouvez lire l'article complet ici (traduction ci-dessous).
Merci à tous les bénévoles qui ont soutenu FightAIDS@Home, OpenPandemics - COVID-19, et d'autres projets pour la recherche de nouveaux et meilleurs traitements.
La suite AutoDock à 30 ans
David S. Goodsell 1,2, Michel Sanner 1 Arthur J. Olson 1 , Stefano Forli 1
1. Département de biologie structurelle et computationnelle intégrative, The Scripps Research Institut, La Jolla, CA USA
2. Collaboratoire de recherche pour la banque de données de protéines de bio-informatique structurelle, Rutgers, The State Université du New Jersey, Piscataway, NJ USA
titre courant : La suite AutoDock à 30 ans
Auteurs correspondants : David S. Goodsell, Stefano Forli
L'institut de recherche Scripps
10550 N Torrey Pines Road
La Jolla, CA 92037Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser. ,Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La suite AutoDock fournit un ensemble complet d'outils pour l'arrimage moléculaire par ordinateur des ligands et la conception et le développement de médicaments. La suite s'appuie sur 30 ans de développement de méthodes, notamment les champs de force empiriques à énergie libre, les moteurs d'arrimage moléculaire, les méthodes de prédiction des sites et les des outils de visualisation et d'analyse. Des outils spécialisés sont disponibles pour les systèmes difficiles, y compris les inhibiteurs covalents, les peptides, les composés à macrocycles, les systèmes où ils sont ordonnés. l'hydratation joue un rôle clé, et les systèmes avec une grande flexibilité de récepteurs. Toutes les méthodes d'AutoDock sont disponibles gratuitement pour l'utilisation et la réutilisation, ce qui a engendré la poursuite de la la croissance d'une communauté diversifiée d'utilisateurs principaux et de développeurs tiers.
Énoncé général : L'arrimage moléculaire par ordinateur est largement utilisé pour étudier les propriétés de liaison des ligands aux molécules biologiques et pour la découverte et la conception de nouveaux médicaments sur la base de leur structure. La suite AutoDock comprend une collection complète d'outils de calcul pour la préparation, l'exécution, la visualisation et l'analyse des expériences d'arrimage moléculaire par ordinateur. La suite AutoDock est librement disponible et a été utilisé par une communauté diversifiée d'utilisateurs et de développeurs.
Introduction
AutoDock a été présenté il y a trente ans comme la première méthode d'arrimage moléculaire des ligands flexibles à des protéines. À l'époque, les méthodes de prédiction de la structure biomoléculaire étaient souvent strictement limitées par l'état naissant de l'infrastructure informatique, qui était limitée à la fois en termes d'espace de stockage et la vitesse de calcul. De puissantes méthodes de dynamique moléculaire telles que AMBER 1 ont permis des représentations basées sur la physique, mais ils étaient limités à de courtes échelles de temps et exploraient généralement des espaces conformationnels autour d'un modèle de départ. La méthode révolutionnaire DOCK utilisait une représentation simplifiée des interactions ligand-récepteur, avec des ligands rigides et une méthode basée sur le score des champs d'interactions, et a ainsi pu rechercher les espaces conformationnels les plus importants nécessaires aux études d'arrimage moléculaire informatique et à la découverte de médicaments. AutoDock a pris une décision intermédiaire en utilisant un champ de force basé sur la physique, similaire à celui de l'AMBRE, mais en l'utilisant avec une approche volumétrique rapide d'évaluation énergétique pour permettre un amarrage qui explore les grands espaces conformationnels d'un ligand flexible.
Trente ans plus tard, la suite AutoDock a été utilisée dans de nombreuses recherches et des efforts de médecine dans le monde entier. En cherchant "AutoDock" dans PubMedCentral, on obtient plus de 7000 résultats et un total combiné de 30 000 citations sont signalées par Google Scholar pour les trois publications les plus citées de la suite AutoDock. Il s'agit notamment de rapports sur l'amarrage moléculaire, la méthodologie de conception des médicaments et les principales applications. Pour se faire une idée de la diversité de ces applications, nous avons fait une enquête sur les candidatures publiées dans le JACS à partir de la liste PubMedCentral, trouver des études portant, par exemple, l'analyse de substrats non naturels de la strictosidine synthase, la liaison du ligand dans une streptavidine artificielle Rh(III) métalloenzyme, la liaison des intercalaires de l'ADN covalent, la caractérisation de la liaison des colorants aux oligomères solubles des amyloïdes Aβ, l'évaluation des inhibiteurs covalents ciblés à partir d'un effort de découverte de ligands à base de fragments pour cibler un protéome de cellule de cancer du colon et le dépistage virtuel des glycanes contre la sialoadhésine.
Aujourd'hui, les utilisateurs disposent de multiples options académiques et commerciales pour l'amarrage moléculaire (voir, par exemple, plusieurs examens récents), et des copies d'écrans d'amarrage moléculaire virtuelles de plus en plus nombreux sont régulièrement effectués avec des résultats probants. Toutefois, l'efficacité de l'arrimage moléculaire et du dépistage virtuel reste limitée par les défis posés à la fois par l'interaction des utilisateurs et par les simplifications physiques nécessaires. Pour relever ces défis, le développement d'AutoDock se poursuit avec deux objectifs à l'esprit. Tout d'abord, une voie de développement fortement axée sur l'utilisateur a produit des outils validés pour une utilisation générale, y compris les interfaces graphiques, les modules d'ancrage et les outils d'analyse destinés à un large éventail d'experts et d'utilisateurs non experts. Deuxièmement, nous avons poursuivi de multiples voies parallèles de développement de nouvelles méthodes, en utilisant l'approche AutoDock comme un rat de laboratoire pour remédier aux limites actuelles des méthodes d'amarrage, y compris les lacunes dans les méthodes de notation et l'extension des recherche dans des espaces chimiques de plus en plus grands (figure 1).
La suite AutoDock
Les méthodes d'arrimage moléculaires informatique cherchent à prédire l'interaction entre les ligands et les cibles de protéines macromoléculaires. L'amarrage moléculaire est généralement utilisé dans le cadre d'une plus large exploration ou d'une ligne de conception (figure 2). Deux problèmes sous-jacents doivent être résolus dans toute méthode d'amarrage moléculaire efficace. Tout d'abord, un champ de force est requis pour marquer des poses d'essai du complexe, en espérant qu'elles reflètent les énergies sous-jacentes de l'interaction biomoléculaire. Deuxièmement, une méthode de recherche est nécessaire pour explorer suffisamment d'espace conformationnel d'interaction disponible afin de garantir qu'une réponse est obtenue. Au début du développement de la suite AutoDock, nous avons pris la décision de construire nos champs de force sur une base solide de méthodes basées sur la physique, qui ont montré beaucoup de succès dans la prédiction de la structure, des interactions et des propriétés biomoléculaires. Le défi a été de simplifier ces champs de force de manière à permettre leur utilisation dans les différentes méthodes de recherche nécessaires pour explorer les grands espaces conformationnels d'arrimage moléculaire.
La première version d'AutoDock combinait une approche volumétrique de l'évaluation énergétique avec une méthode tempérée de recherche simulée. Plusieurs approximations ont été nécessaires pour permettre l'amarrage moléculaire dans des temps raisonnables sur les niveaux VAX des ordinateurs de l'époque (figure 3). Des cartes volumétriques sont pré-calculées pour chaque type d'atome de ligand en balayant le scan des atomes examinés à travers l'espace occupé par la cible, avec pour conséquence qu'ils imposent une limitation d'un récepteur. En outre, les degrés de liberté conformationnels du ligand ont également été limités à des rotations de torsion, avec des longueurs et des angles de liaison contraints à la géométrie de la pose du départ. Cela repose sur l'hypothèse que la conformation liée est une variation en torsion de la conformation des entrées. Les limitations de l'espace de stockage ont également nécessité la réduction du nombre d'atomes et donc le nombre et la taille des cartes à calculer et à stocker. La suite AutoDock s'est développée à partir de cette base, fournissant actuellement de multiples méthodes d'amarrage moléculaire, des interfaces graphiques d'utilisateur et des outils d'analyse (tableau 1).
La version actuelle d'AutoDock, AutoDock4 (AD4), conserve une grande partie des concepts originaux d'évaluation de l'énergie et a nettement amélioré les capacités de recherche, en augmentant la complexité des ligands qui peuvent être arrimés moléculairement. Les améliorations du champ de force ont porté sur l'amélioration de la géométrie de la liaison hydrogène et la pondération empirique des paramètres du champ de force pour prévoir des pliages d'énergies libres, et les développements en cours décrits plus en détail ci-dessous. La collaboration avec les informaticiens a permis la mise en œuvre d'un algorithme génétique hybride/méthode de recherche local qui étend considérablement la portée de la recherche conformationnelle. Des améliorations récentes comprennent l'ajout de gradients à la description du champ de force et le portage de l'AD4 sur des unités de traitement graphique (GPU) pour améliorer encore les performances.
AutoDockVina (ADVina) est une méthode d'amarrage moléculaire clé en main qui a permis de faire le point sur l'état de l'art en 2010 en amarrage moléculaire. De nombreuses optimisations sont utilisées pour améliorer la vitesse d'amarrage moléculaire, notamment une fonction de notation par morceaux qui se prête à une évaluation rapide et qui a été calibrée en utilisant ~1300 complexes de PDB-Bind, et une méthode de recherche hautement optimisée basée sur un modèle de Monte-Carlo et l'optimisation locale basée sur les gradients. Une grande partie du mécanisme d'arrimage moléculaire qui est exposée dans les programmes AutoDock, comme l'utilisation de cartes pour l'évaluation énergétique, est cachée dans ADVina, ce qui en fait une bonne option à utiliser par des non-experts.
AutoDockFR (ADFR, "Flexible Receptor") est un effort de développement parallèle qui porte sur la limitation d'un récepteur rigide, s'appuyant sur le programme FLIPDock précédent comprenant la capacité à modéliser explicitement des chaînes latérales flexibles définies par l'utilisateur, mais l'ADFR crée une représentation du récepteur, permettant de définir les chaînes latérales, les boucles et les domaines subissant des mouvements lors des recherches conformationnelles. L'ADFR met en œuvre un algorithme génétique efficace qui permet de spécifier jusqu'à 15 chaînes latérales flexibles dans le site de liaison du récepteur. Il permet également de fixer des ligands covalents et de contraindre les atomes des ligands à des positions prédéfinies en utilisant les potentiels harmoniques. S'appuyant sur ces travaux, AutoDock CrankPep (ADCP) est un moteur d'amarrage moléculaire développé spécifiquement pour l'amarrage des peptides. L'ADCP combine l'échantillonnage de conformation des peptides Crankite avec les cartes d'affinité AutoDock pour un arrimage moléculaire efficace des peptides linéaires ou cycliques avec jusqu'à 20 acides aminés à partir de leur séquence.
La disponibilité du logiciel libre et la conception modulaire de la suite AutoDock ont également favorisé l'utilisation par de nombreux développeurs tiers. Citons par exemple Smina, une branche d'ADVina qui rationalise la personnalisation des champs de force, et PSO@AutoDock, une mise en œuvre de l'optimisation des essaims de particules dans AutoDock3.
Les avantages et les inconvénients des cartes
Le pré-calcul volumétrique des énergies d'interaction était l'innovation centrale qui a rendu possible l'amarrage moléculaire flexible dans les versions initiales d'AutoDock, et il reste essentiel pour réduire la complexité informatique du problème d'amarrage moléculaire. Dans le cas idéalisé d'un "verrouillage" parfait de l'interaction sous sa forme liée, définie et maintenue en place par des interactions spécifiques et stériques avec le récepteur voisin. En réalité, cependant, les macromolécules biologiques sont composées d'atomes de taille finie, de sorte que la forme du site actif est le résultat d'un compromis évolutif entre la nécessité de créer cette signature parfaite et contraignante, mais limitée par le produit chimique et les propriétés physiques de la protéine ou du polymère d'acide nucléique. Par conséquent, ces cartes nécessitent typiquement une certaine intuition chimique pour l'interprétation.
Néanmoins, les cartes d'interaction sont des outils puissants dans l'arsenal des méthodes de découverte de médicaments
et de la conception. S'appuyant sur l'idée que l'image latente des ligands préférentiels est contenue dans ces cartes, nous avons mis au point deux méthodes de prévision des sites de liaison. AutoLigand combine des cartes pour les atomes de carbone et les liaisons des atomes d'hydrogène, créant ainsi une carte combinée qui détermine le meilleur type d'atome pour chaque endroit. Ensuite, l'utilisateur définit une taille souhaitée pour le ligand et trouve le meilleur ensemble de points contigus dans les cartes ayant un tel volume.
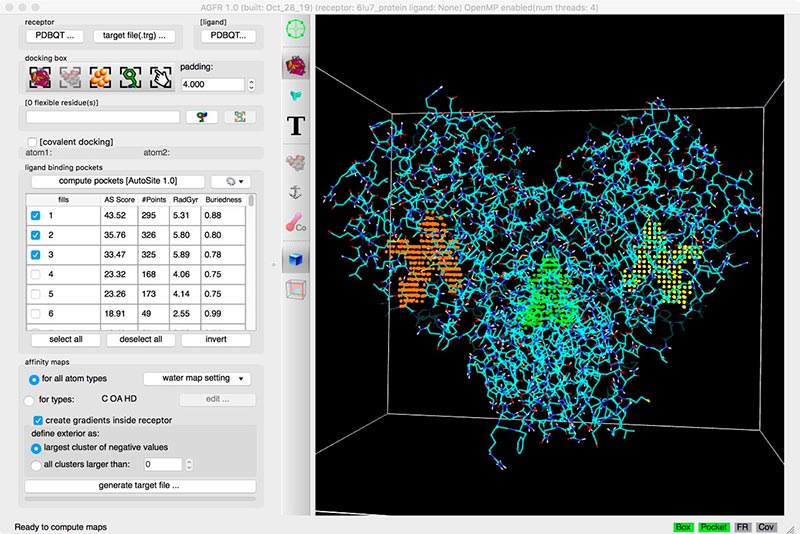
AutoSite (Figure 4) adopte une approche légèrement différente, en localisant ces sites de liaison idéaux par regroupement des régions de haute affinité. Les deux méthodes peuvent être utilisées pour identifier les sites de liaison propices et pour caractériser la forme optimale des ligands qui se lieront aux sites.
Au fil des ans, nous avons également expérimenté des cartes spécialisées pour des applications particulières.
Par exemple, une simple carte qui indique la distance jusqu'à l'atome le plus proche peut être utilisée pour mettre des ligands en contact avec des protéines, réduisant ainsi l'exploration inutile de conformations complètement entourées de solvant. De petits niveaux de mouvement de la protéine peuvent être accommodés pendant l'amarrage moléculaire grâce à l'utilisation de cartes "lissées" qui évaluent l'énergie minimale dans un seuil de faible distance de chaque point, ou en créant des cartes qui combinent les contributions de multiples conformations de la protéine. Une approche plus directe peut également être adoptée, appelée "méthode des complexes relaxés", dans laquelle des instantanés d'une protéine sont pris à partir de la dynamique moléculaire, puis utilisés dans des expériences d'amarrage moléculaire individuelles. L'application d'AutoDock dans un ensemble d'arrimage
a joué un rôle dans le développement du premier inhibiteur de l'intégrase du VIH-1 cliniquement approuvé par Merck 3.
Interfaces graphiques utilisateur
Il est absolument essentiel de disposer d'un système frontal réactif pour aider les utilisateurs à tracer leur chemin dans leurs applications. AutoDockTools (ADT), basé sur les méthodes graphiques modulaires de MGLTools, a servi à cette fin pour les différentes versions d'AutoDock. Il fournit des outils graphiques pour ajouter des hydrogènes et définir l'articulation des ligands, préparer les parties flexibles et rigides des récepteurs, créer des fichiers de commande et enfin analyser les résultats des simulations d'amarrage moléculaire. Toutefois, ADT est un outil conçu en grande partie pour les utilisateurs ayant une connaissance approfondie de la modélisation moléculaire et des méthodes d'amarrage moléculaire, avec une grande partie de la machinerie d'AD4 exposée pour permettre la personnalisation pour des applications difficiles. En outre, il est principalement utile pour la spécification d'un petit nombre d'expériences d'amarrage moléculaire, et est lourd lorsqu'il est appliqué à des problèmes plus importants comme le dépistage virtuel. Ces limitations ont été et sont encore prises en compte par de nouveaux outils.
Chimera 35, PyMOL 36 et de nombreux autres outils tiers ont intégré la possibilité de spécifier et d'écrire des fichiers de commande pour ADVina, offrant ainsi une approche clé en main de l'amarrage pour les non-experts.
Nous sommes en train de développer un frontal clé en main similaire, qui fournit une interface de type "pointer-cliquer" permettant aux utilisateurs de personnaliser et d'organiser les coordonnées des ligands et des récepteurs à partir des entrées des archives PDB, et de spécifier des simulations d'amarrage moléculaire avec les choix résultants (voir ci-dessous).
Nous avons développé Raccoon comme un outil permettant de spécifier et de gérer des écrans virtuels avec la suite AutoDock. Il utilise une approche de base de données flexible pour préparer et gérer de grandes bibliothèques de ligands, dispose de mécanismes pour lancer et contrôler les simulations d'amarrage moléculaire sur les groupes de calcul et, surtout, dispose d'un arsenal d'outils de filtrage flexibles pour analyser les résultats et isoler les composés prometteurs pour des études supplémentaires (figure 5).
Améliorations
La biologie étant la biologie, il y a des exceptions à toutes les règles. La fonctionnalité de base de la suite AutoDock a été créée pour être un outil général, paramétré et validé par rapport à un ensemble diversifié de ligands de type médicamenteux se liant à des sites de liaison de protéines de poche. Entre nos mains, ces méthodes de base donneront des résultats d'arrimage clés en main pour environ la moitié des nouveaux systèmes d'essai. Dans d'autres cas, la biologie impose de nouveaux aspects qui ne sont pas efficacement traités dans la suite principale, de sorte que nous avons passé la majeure partie des trois dernières décennies à créer une série d'améliorations pour relever ces défis.
Les effets des solvants restent l'un des plus grands défis à relever pour améliorer la précision et la spécificité des simulations d'amarrage moléculaire. AD4, ADVina et ADFR intègrent tous des approches empiriques pour estimer les conséquences énergétiques de la désolvabilité, basées sur des mesures qui se rapprochent de la quantité d'eau déplacée par un ligand lorsqu'il se lie. Ces méthodes utilisent des fonctions avec une très faible dépendance à la distance et ne tiennent pas compte des effets localisés de l'eau de pontage. Nous avons développé une approche plus explicite de ce problème en attachant les eaux à toutes les positions possibles d'interaction sur un ligand, puis en permettant à ces eaux d'interagir avec les protéines ou de disparaître pendant la simulation d'amarrage moléculaire (figure 1).
Des potentiels spécialisés peuvent être utilisés pour incorporer des liaisons covalentes dans les simulations d'amarrage moléculaire.
Ces dernières peuvent être abordées de plusieurs manières. Des potentiels internes peuvent être ajoutés pour créer des liaisons au sein des ligands. Cela a été utilisé avec succès pour modéliser des ligands avec des macrocycles flexibles : l'anneau est brisé à un endroit, et la simulation d'amarrage moléculaire est effectuée avec un potentiel personnalisé qui favorise la reconstitution de la liaison macrocyclique d'origine. De même, en s'appuyant sur des travaux antérieurs sur l'arrimage moléculaire covalent, nous avons développé notre approche la plus efficace pour la prédiction et la conception d'inhibiteurs covalents ciblés. Appelée "reactive docking", la méthode utilise un potentiel personnalisé entre le ligand et le récepteur pour évaluer la capacité des ligands à se lier à des sites sur la protéine et forment ensuite des liaisons covalentes avec les sites adjacents de la réaction chimique (figure 6).
Nous avons conçu des méthodes permettant d'exploiter des informations expérimentales ou dérivées d'une autre manière pour orienter les calculs vers l'établissement d'interactions précises entre le ligand et la protéine. Par exemple, l'analyse de la dynamique moléculaire du cosolvant a été utilisée pour identifier les points chauds de liaison à la surface de la protéine et orienter les positions du ligand pour établir des interactions spécifiques avec ces régions. De même, l'ADFR prend en charge les contraintes harmoniques qui pénalisent les atomes de ligands spécifiés par l'utilisateur qui s'éloignent de la position prédéfinie, permettant ainsi un "ancrage", où un groupe d'atomes de ligands est soumis à une telle contrainte. Elle met également en œuvre le concept de
la recherche de voisinage, où la recherche d'amarrage moléculaire se concentre sur l'exploration du "voisinage" d'une position initiale accostée, le voisinage étant défini par une coupure RMSD.
Le champ de force de base de la suite AutoDock est paramétré et optimisé pour des types d'atomes standard, lorsqu'ils sont disposés en molécules standard semblables à des médicaments. Plusieurs laboratoires ont effectué des paramétrages personnalisés pour des systèmes spécifiques, comme une approche optimisée pour la liaison des glucides aux protéines. Nous avons concentré notre attention sur les aspects du champ de force qui manquent, comme les nouveaux potentiels pour spécifier la coordination par des ions métalliques tels que le zinc. Nous avons également lancé un effort pour améliorer les potentiels utilisés pour spécifier la liaison de l'hydrogène, en commençant par une étude détaillée de la force et de la direction de la liaison de l'hydrogène en utilisant une analyse de mécanique quantique des composés modèles.
Performance et choix du meilleur outil
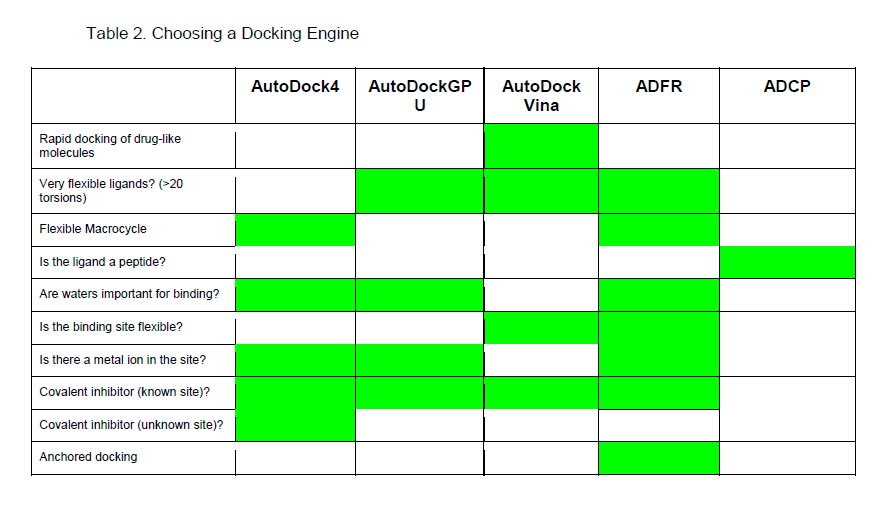
Comme mentionné ci-dessus, nous avons suivi une approche à plusieurs voies pour le développement de la suite AutoDock afin de permettre l'exploration de diverses nouvelles méthodes. Cette approche, ainsi que le fait que de nombreuses autres méthodes d'ancrage académiques et commerciales efficaces sont actuellement disponibles, peuvent rendre difficile le choix d'un outil pour une application particulière. Dans la suite AutoDock, nous proposons généralement ADVina comme approche de première ligne, clé en main, car elle est rapide pour les ligands typiques de type médicamenteux. Pour les systèmes présentant différents défis non conventionnels, tels que la réactivité chimique, la flexibilité des récepteurs ou les systèmes nécessitant des paramétrages ad hoc, d'autres outils de la suite peuvent être appliqués pour les modéliser. Le tableau 2 présente des suggestions d'outils appropriés pour diverses applications courantes.
De nombreuses études de tiers ont été présentées, qui quantifient les performances des méthodes de la suite AutoDock et les comparent avec d'autres outils disponibles. Par exemple, une récente étude détaillée de cinq méthodes commerciales et cinq méthodes académiques avec deux mille complexes de PDBBind a montré une performance comparable d'ADVina, Glide, GOLD, et plusieurs autres, obtenant les meilleures positions amarrées dans environ la moitié des systèmes à 2,0 A RMSD près, et des coefficients de corrélation d'environ 0,5 pour la prédiction de l'énergie. Ces résultats reflètent nos propres conseils aux utilisateurs, basés sur les résultats de nos études de validation : en général, les méthodes d'amarrage réussissent environ la moitié du temps, avec de meilleures statistiques pour les ligands plus petits et moins flexibles et les cibles à flexibilité limitée, et les énergies sont prévues à environ 2-3 kcal/mol près, ce qui permet de séparer les inhibiteurs milli-, micro- et nano-molaires, mais n'est pas efficace pour le classement avec des différences énergétiques plus fines.
Dépistage virtuel avec la suite AutoDock
L'efficacité et les limites des méthodes actuelles d'amarrage moléculaire deviennent plus apparentes dans les efforts de dépistage virtuel. L'état de l'art actuel de la suite AutoDock, et de même pour la plupart des méthodes d'amarrage moléculaire actuelles, fournit des conformations amarrées cohérentes pour des molécules ayant environ une douzaine de degrés de liberté en torsion, avec des énergies libres prévues à environ 2-3 kcal/mol près, dans des systèmes où le mouvement des protéines ne joue pas un rôle significatif. Ce niveau de précision s'est avéré suffisant pour assurer le succès des efforts de criblage virtuel, avec l'espoir qu'entre 1 et 10 % des résultats virtuels prévus se révèlent présenter une affinité de liaison détectable lors des tests expérimentaux.
Le filtrage virtuel est sans doute la principale application de la communauté actuelle des utilisateurs d'AutoDock, et une grande partie des efforts de développement de la dernière décennie ont été axés sur l'amélioration de l'infrastructure et des résultats. Raccoon est le principal frontal supportant les écrans virtuels fourni avec la suite AutoDock (figure 5). Divers services web sont également disponibles auprès de tiers, tels que MtiOpenScreen et DrugDiscovery@TACC
(https://drugdiscovery.tacc.utexas.edu/).
Le développement des champs de force a également permis de relever certains des défis posés par le filtrage virtuel. Par exemple, en 2007, nous sommes passés à un modèle énergétique basé sur un cycle thermodynamique qui comprend une évaluation explicite des états liés et non liés. Des résultats prédictifs légèrement meilleurs ont été obtenus avec un protocole qui estimait les énergies des contacts intramoléculaires dans les ligands libres dans le cadre de ce cycle, mais ce protocole a finalement été abandonné pour un modèle plus simple qui suppose que les effets intramoléculaires sont similaires dans les états liés et non liés du ligand. Le modèle plus complet a systématiquement classé en tête de liste un ensemble de molécules entassées avec des affrontements internes, en raison de l'instabilité informatique des très hautes énergies de la forme non liée.
Le dépistage virtuel a entraîné le besoin de ressources informatiques toujours plus importantes, car plus la botte de foin est grande, plus elle risque de contenir une aiguille dorée. Heureusement, le criblage virtuel peut être "parallèlement embarrassant" en affectant chaque arrimage moléculaire composé à un processeur différent fonctionnant en parallèle, ce qui donne une vitesse pratiquement linéaire. En 2000, en collaboration avec Entropia, une start-up spécialisée dans le calcul, nous avons lancé FightAIDS@Home (FAAH) pour démontrer l'utilité pratique du calcul sur grille dans la conception de médicaments. FAAH a été le premier projet biomédical développé pour une plateforme de volontariat sur grille, faisant suite à des projets antérieurs de science citoyenne comme SETI@Home et le GIMPS (Great Internet Mersenne Prime Search) qui ont utilisé le calcul bénévole pour résoudre des problèmes astronomiques et mathématiques. Avec une telle ressource faisant tourner AutoDock sur des milliers de processeurs largement distribués, nous avons pu étendre nos études de la biologie structurelle du VIH avec de grands écrans virtuels. En 2005, FAAH a rejoint le World Community Grid d'IBM, qui a amplifié les ressources disponibles pour plus de 3 millions de volontaires CPU, augmentant considérablement la taille des bibliothèques de composés recherchés, ainsi que les degrés de liberté dans les modèles des protéines cibles.
Au cours de ces 20 dernières années, l'application de la suite AutoDock (AD4 et ADVina) sur les plateformes FAAH et locales a permis d'informer et d'élargir les approches du développement thérapeutique du VIH. Nous avons examiné le rôle de la flexibilité des protéines dans l'inhibition allostérique de la protéase du VIH et de l'intégrase en utilisant un panel d'instantanés de la dynamique moléculaire ; nous avons évalué de nouveaux inhibiteurs de protéase à large spectre (VIH/FIV), identifié de nouvelles cibles pour le développement de médicaments comme la capside du VIH, et exploré les mécanismes de l'évolution de la résistance du VIH aux médicaments. Grâce à un nombre considérable de résultats d'arrimage moléculaire provenant de grands cribles virtuels FAAH, nous avons utilisé l'apprentissage machine sur les données générées par FAAH pour améliorer les critères de sélection des vrais positifs dans le dépistage. Afin d'affiner encore les résultats des criblages AutoDock avec des calculs plus intensifs d'énergie gratuite, FAAH a maintenant entamé une deuxième phase en collaboration avec le laboratoire Levy, dans laquelle des simulations de dynamique moléculaire sont utilisées pour fournir de meilleures estimations de l'énergie libre de la liaison.
Récemment, en collaboration avec IBM World Community Grid, nous avons lancé le projet OpenPandemics - COVID-19 utilisant AutoDock (https://www.worldcommunitygrid.org/research/opn1/overview.do) pour réaliser de grands écrans virtuels contre de multiples sites cibles dans le protéome du SRAS-CoV-2 afin de rechercher de nouveaux candidats thérapeutiques COVID-19 potentiels, y compris des inhibiteurs covalents réactifs. Outre nos propres efforts, nos moteurs d'amarrage moléculaire soutiennent d'autres projets du World Community Grid qui réalisent des criblages virtuels ciblant le cancer, la malaria et le virus Ebola. L'ampleur de ces efforts de calcul distribué pose des défis uniques en matière de gestion et d'analyse des données d'ancrage qui sont abordés dans nos efforts de développement actuels.
L'amarrage moléculaire des peptides
Depuis la première version d'AutoDock, les utilisateurs ont repoussé les limites de ce qui peut être efficacement modélisé avec l'arrimage moléculaire informatique. Les peptides, en particulier, ont suscité un intérêt constant de la part de la communauté des utilisateurs, mais qui nécessite une approche créative. La motivation est claire : les peptides thérapeutiques ont connu une renaissance ces dernières années, comme en témoignent les 60 médicaments à base de peptides actuellement approuvés sur les principaux marchés, et plus de 150 en développement clinique actif. Les peptides cycliques présentent un intérêt particulier, avec 40 exemples en usage clinique actuel et en moyenne un nouveau qui arrive sur le marché chaque année. L'amarrage moléculaire des peptides, cependant, présente des défis de taille, tant pour la recherche que pour la notation.
La taille de l'espace-solution à explorer lors de l'amarrage moléculaire croît de manière exponentielle avec chaque variable ajoutée à optimiser. Les méthodes de recherche développées pour les petites molécules de type médicamenteux, qui ont généralement une douzaine de liaisons rotatives, ont donc peu de chances d'échantillonner correctement les 50 à 100 liaisons rotatives des peptides. Les peptides ont également tendance à s'associer à leurs partenaires de liaison dans des sillons peu profonds à la surface, tandis que les molécules de type médicamenteux se lient plus souvent dans des poches profondes propices à l'amarrage moléculaire qui minimisent leurs interactions avec le solvant. Par conséquent, les fonctions de notation développées et calibrées pour les petites molécules médicamenteuses sont souvent peu performantes pour les peptides, même les plus courts.
Les premiers travailleurs ont résolu ces problèmes en divisant les peptides en fragments, en les amarrant séparément, puis en choisissant des positions qui pouvaient être recombinées en une séquence peptidique souhaitée. Étonnamment, même l'amarrage protéine-protéine se prête à cette approche. Nous nous attaquons actuellement à ces problèmes en explorant des représentations simplifiées de l'espace conformationnel basées sur les transformations de la rotation du pivot (figure 3). L'ADCP, qui permet actuellement l'amarrage moléculaire cohérent de peptides comportant jusqu'à 20 acides aminés (figure 7), est à la pointe de ce domaine dans une évaluation récente de 14 programmes d'amarrage de peptides. L'ADCP peut également cycliser les peptides tête-bêche (figure 7.A) et/ou en formant jusqu'à 2 ponts disulfure lorsque les cystéines sont présentes (Fig 7.B), soutenant ainsi des molécules à cycles multiples. La cyclisation est réalisée à la volée pendant la simulation d'amarrage moléculaire en utilisant des potentiels pour tirer les atomes de soufre N et C-terminaux ou de cystéine ensemble tout en ignorant la répulsion stérique entre ces atomes. Dans ce dernier cas, l'appariement des cystéines n'a pas besoin d'être spécifié par l'utilisateur, mais résulte plutôt de l'amarrage moléculaire.
Développement actuel et orientations futures
Si l'on examine le vaste ensemble de recherches et de développements qui citent les publications d'AutoDock, et notre correspondance permanente avec les utilisateurs, on constate que nos principaux utilisateurs sont généralement des travailleurs dans des domaines connexes - biochimistes, biologistes structurels, physiciens - qui n'ont pas de compétences approfondies en matière de chimie computationnelle et d'arrimage moléculaire. Pour cette communauté, la suite AutoDock, et ADVina en particulier, est un ensemble attrayant étant donné qu'il est gratuit, facilement disponible et relativement simple à démarrer. Cela a donné lieu à des centaines de rapports dans lesquels l'arrimage moléculaire est utilisé pour compléter des études de structure/fonction plus importantes. L'un des principaux objectifs de notre développement actuel et futur est de soutenir cette communauté d'utilisateurs, vaste et croissante. Nous travaillons actuellement à la mise au point d'un frontal graphique unifié qui permettra d'exploiter la facilité d'utilisation d'ADVina, mais aussi d'accéder clé en main aux niveaux de fonctionnalités supplémentaires disponibles dans les autres outils de la suite. Ce frontal intégrera des outils agile pour gérer les nombreux défis posés par les coordonnées d'entrée expérimentales : construction des boucles manquantes et des résidus, protonation et tautomérisation, modèles de charge, flexibilité des récepteurs de manipulation, et des dizaines d'autres obstacles, petits mais essentiels (figure 8). Le frontal fournira également des outils faciles pour gérer les expériences d'amarrage moléculaire, allant de simples études d'un ligand d'essai avec un récepteur à des écrans virtuels, ainsi que des outils d'analyse pour filtrer et interpréter les résultats. Enfin, le frontal comprendra une fonctionnalité complète pour saisir la provenance de chaque expérience, garantissant la reproductibilité du travail effectué au sein de la suite. Nous espérons que cette interface fournira à terme un accès principal à la suite AutoDock pour tous les niveaux d'expertise des utilisateurs. Nous développons actuellement des composants enfichables de cette interface qui feront partie du frontal que nous envisageons, offrant un chemin progressif vers l'objectif ultime d'un environnement unifié où les utilisateurs peuvent facilement accéder à tous les outils développés dans nos laboratoires.
Nous bénéficions également d'une communauté dynamique et créative de développeurs tiers, et en réponse, nous avons cultivé une approche ouverte du développement de la suite AutoDock pour soutenir l'extension et l'innovation de cette importante communauté d'utilisateurs. Tous les composants de la suite AutoDock sont disponibles sous licence open source et accessibles via le site web AutoDock. Ils sont implémentés dans les langages de programmation C, C++ et Python. La suite AutoDock est un vaste écosystème de logiciels composé de nombreux composants logiciels, dont beaucoup peuvent être utilisés indépendamment. Le code source et l'exécutable de ces composants logiciels sont disponibles dans des dépôts indépendants. Bien qu'il soit possible de fournir tous les composants logiciels dans un seul dépôt, le fait de les garder séparés favorise leur indépendance : c'est-à-dire que les utilisateurs téléchargent la source d'un composant logiciel donné, le construisent et l'utilisent sans que les autres soient présents. Si une dépendance est introduite par inadvertance, ce processus échouera et nous alertera de la nouvelle dépendance introduite.
Dans nos propres laboratoires et dans de nombreux autres, les principes de base de l'amarrage moléculaire et de l'évaluation de l'énergie sont découverts et développés. Nos propres travaux se concentrent actuellement sur l'amélioration des fonctions de notation afin de fournir une évaluation et un classement plus précis de l'énergie, y compris l'évaluation des approches d'apprentissage machine pour le réglage ou la mise en œuvre de ces fonctions, et l'extension des méthodes de recherche, les peptides étant un point d'intérêt particulier actuellement. Nous avons travaillé sous le capot pour rendre les méthodes de la suite AutoDock modulaires et extensibles afin de soutenir les travaux qui continueront à étendre la fonctionnalité et la précision des méthodes d'amarrage moléculaire. Nous espérons que la suite AutoDock continuera à bénéficier d'améliorations dans ce domaine, en particulier de la précision de la modélisation rendue possible par l'augmentation spectaculaire de la puissance de calcul ces dernières années. Grâce à l'énorme impulsion donnée par les architectures accélérées telles que les GPU, il sera possible d'effectuer des évaluations énergétiques plus coûteuses et plus précises, en surmontant certaines des limites imposées par le calcul dans les premiers temps. Les outils de la suite AutoDock ont été et continueront à être disponibles gratuitement grâce à une licence open source sur le site AutoDock (site web : https://ccsb.scripps.edu/), avec documentation et tutoriels. Des protocoles détaillés pour l'exécution d'applications communes dans AD4 et ADVina ont été présentés.
Remerciements
Nous remercions Diogo Santos-Martins et Giulia Bianco pour leur aide dans la réalisation du manuscrit. Au cours des 30 dernières années, le développement d'AutoDock a été continuellement soutenu par le NIGMS-NIH, et plus récemment par les subventions GM069832 (SF) et GM096888 (MS). Les auteurs n'ont aucun conflit d'intérêt à déclarer. Il s'agit du manuscrit n° 30005 du Scripps Research Institute.
Figure 1.
Résultat de l'amarrage moléculaire du D3R Grand Challenge 4, avec un ligand macrocyclique lié à BACE1, en utilisant une nouvelle approche d'amarrage moléculaire accéléré par le GPU et un nouveau modèle de ligand hydraté. Tous les sites possibles sur le ligand sont hydratés avec une géométrie idéale (sphères cyan), et après l'amarrage moléculaire, tous sauf un se chevauchent avec la protéine (surface jaune) et sont utilisés pour évaluer une contribution de désolvabilité à l'énergie libre. La pose cristallographique est représentée en vert.
Image générée dans Python Molecule Viewer, y compris AutoDockTools.Figure 2.
Un ligne de conception d'amarrage moléculaire typique commence par les coordonnées d'un récepteur et d'un ligand, à partir de déterminations expérimentales de structure, de modélisation d'homologie, de génération idéalisée à partir de SMILES, etc., présentés ici schématiquement en haut. Les récepteurs et les ligands sont traités pour se conformer à la représentation utilisée dans la méthode d'amarrage moléculaire (attribution des types d'atomes et des charges, définition des modes de flexibilité, etc.), et souvent un site de liaison préféré sur le récepteur est identifié. Le moteur d'amarrage moléculaire prédit alors les positions énergétiquement favorables du ligand dans le site de liaison du récepteur. Dans le criblage virtuel, tout ce processus se déroule à une plus grande échelle, en préparant et en fixant une bibliothèque entière de ligands, puis en filtrant les résultats pour identifier les meilleurs candidats pour une étude plus approfondie.Figure 3.
De nombreuses simplifications sont imposées pour améliorer la vitesse d'accostage.
A) Les énergies d'interaction pour les atomes de la sonde sont calculées de manière volumétrique. Ici, les emplacements favorables pour le carbone (contours blancs) et l'oxygène (contours rouges) sont calculés dans le site actif d'une protéase du VIH résistante aux médicaments (entrée PDB 2hc0). Remarquez les deux lobes de la densité du carbone correspondant aux sites P1 et P1', et l'emplacement favorable de l'oxygène entre les volets, correspondant à l'emplacement d'une eau ordonnée dans la plupart des structures.
B) Seuls les degrés de torsion des ligands (ici, un métabolite monophosphate du remdesivir) sont recherchés, et des types d'atomes limités sont utilisés (ici, des carbones aliphatiques en gris et des carbones aromatiques en vert, et seulement des hydrogènes polaires).
C) Des mouvements plus grossiers sont utilisés pour simplifier la recherche conformationnelle de ligands plus grands, comme les mouvements du vilebrequin pour les peptides.
Images générées dans Python Molecule Viewer, y compris AutoDockTools.
Figure 4.
Prédiction de poche en un clic avec AutoSite dans l'interface graphique utilisateur de l'ADFR. Les trois poches de la protéase majeure SARS-CoV-II (entrée PDB 6lu7) sont représentées, y compris une cavité à l'interface du dimère (vert) et les deux sites actifs (jaune et orange).Figure 5.
Analyse de criblage virtuel à l'aide de Raccoon2 : mise en place d'une petite bibliothèque de ZINC 62 sur la kinase c-Abl, montrant la conformation de l'imatinib (entrée PDB 1iep) redécouplée, utilisée comme composé de référence.Figure 6.
Méthodes de liaison covalente supportées dans l'AD4.
A) La méthode de l'attracteur à deux points utilise deux cartes spécialisées (X et Z) qui ont des énergies favorables au site de fixation sur la protéine, entraînant l'amarrage du ligand à l'endroit de la liaison covalente.
B) La méthode d'amarrage réactif utilise un potentiel personnalisé qui conduit l'atome réactif du ligand (R) à la position de quasi-attaque sur l'acide aminé cible.
C) L'approche liée utilise la méthode de la chaîne latérale flexible pour optimiser une position arbitraire du complexe covalent.Figure 7.
Panneau supérieur : meilleure solution d'ancrage (cyan) et structure cristalline (magenta) d'un peptide linéaire qui forme un interrupteur auto-inhibiteur dans la formine mDia1 (entrée PDB 2f31). En partant de la séquence, ADCP plie et place le peptide dans le récepteur. Les interactions entre la chaîne latérale et le récepteur sont bien prévisibles, sauf pour E2, E11 et R20 qui trouvent des plaques polaires.
Panneau inférieur : épine dorsale des peptides cycliques amarrés (boule et bâtonnet) et structure cristalline (réglisse).
(A) La pose amarrée la mieux classée pour une protéine adaptateur de substrat d'ubiquitin ligase avec un peptide cyclique modifié provenant de l'une de ses cibles (entrée PDB 3zgc). Les résidus sont colorés de bleu à rouge pour indiquer un enregistrement correct.
(B) Résultat de l'enregistrement d'un peptide lié à un disulfure interne du virus Epstein Barr affiché par le CMH (entrée PDB 5grd). Les résidus sont codés par couleur selon le type de résidu et les chaînes latérales des deux cystéines montrent la liaison disulfure créée.Figure 8.
Interface utilisateur graphique en cours de développement pour la préparation des molécules à l'amarrage moléculaire, représentée ici avec un mutant cancéreux p53 (entrée PDB 6ggd). Lorsque le fichier PDB est chargé, le contenu moléculaire est analysé, classé et présenté dans un widget d'arbre permettant la spécification des résidus qui seront inclus dans le récepteur ou le(s) ligand(s). Toutes les biomolécules sont disponibles ainsi que l'unité asymétrique. Les acides aminés standard avec des atomes de chaîne latérale manquants sont sélectionnés pour la reconstruction et un rotamer par défaut est sélectionné (ici, Arg290 et Lys291). D'autres emplacements sont affichés et, par défaut, celui qui est le plus occupé est sélectionné (par exemple, Leu252@A). Le ligand de la structure cristalline est marqué pour être préparé en tant que ligand pour l'amarrage. Les segments d'acides aminés manquants sont affichés (en orange). Le widget de l'arbre est lié à la vue 3D, ce qui permet à l'utilisateur de se concentrer sur les zones potentiellement problématiques et d'inspecter la solution proposée.

14 sept. 2020
traduction de l'article WCG : https://www.worldcommunitygrid.org/about_us/viewNewsArticle.do?articleId=645&messageId=175292.1002.1600178732444