


|
|
|
Projet du laboratoire Backer, département de Biochimie de l'Université de Washington (Malaria, Anthrax, VIH, Alzheimer, Cancer de la Prostate ,...) |



URL du projet : http://boinc.bakerlab.org/rosetta/
Détail technique : lien pour rêgler la durée d'une unité de calcul. Appuyer sur Edit Rosetta@home preferences. Target CPU run time. Choisir dans le menu déroulant la durée qui vous convient, ça va de 1 heure à 24h. Sauvegarder en appuyant sur Update preferences.
Par défaut, une unité de calcul dure 6 heures.
Mémoire vive recquise : 512 Mo
|
Liens du Projet
|
L'Alliance Francophone
|
Statistiques
|
Résultats et divers
|
|
-
Les résultats : Domaine Pubic
-
Début du projet : 26 Juin 2005
Contre quoi rosetta@home aide à lutter
Page d'origine de cette traduction
Commentaires de David Baker
Mon groupe de recherche est impliqué aussi bien dans la recherche fondamentale que dans la lutte directe contre les maladies. La plupart des informations sur ce site se concentrent sur la recherche basique, mais j'ai pensé que vous pourriez être intéressés par notre travail sur certaines maladies.
Prédiction et Conception de Structures et Interactions Macromoléculaires
Page d'origine de cette traduction
Ecran de Veille
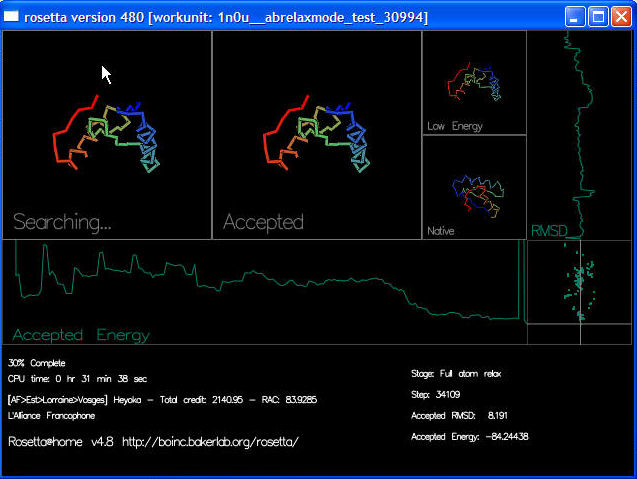
Explication en détails de l'écran de veille
Présentation du projet par David Baker et son équipe (sous-titrage en français)
-
Malaria : Nous collaborons avec un projet mené par Austin Burt du Collège Impérial de Londres qui est une des composante du projet "Grands défis pour la santé mondiale". La malaria est provoquée par un parasite qui passe une partie de son cycle de vie dans l'organisme d'un moustique, et qui est ensuite transmit aux humains par une simple piqure. L'idée du projet est de rendre des moustiques résistants en éliminant de son organisme les gènes indispensables à la survie du parasite. Notre rôle dans le projet est d'employer nos méthodes de modélisation sur ordinateur (ROSETTA) pour concevoir de nouvelles enzymes qui viseront spécifiquement ces gènes en les inactivant
- Anthrax : Nous utilisons ROSETTA pour aider le groupe de recherche de John Collier d'Harvard à établir des modelisation de la toxine d'anthrax qui devraient contribuer au développement de traitements. Vous pouvez lire l'extrait d'un article décrivant une partie de ce travail sur : http://www.pnas.org/cgi/content/abstract/102/45/16409 .
- VIH : Une des raisons qui rend le virus du VIH si mortel est qu'il a évolué pour duper le système immunitaire. Nous collaborons avec des chercheurs à Seattle et au NIH pour essayer de développer un vaccin contre le VIH. Le rôle que nous occupons dans ce projet est central, nous employons ROSETTA@home pour concevoir de petites protéines qui mettront en évidence le petit nombre de régions critiques de la protéine d'enveloppe du VIH , de ce fait le système immunitaire peut facilement identifier et produire des anticorps. Notre but est de créer des vaccins à partir de petites protéines stables qui peuvent être fabriquées à bas prix et diffusées dans le monde entier.
- La maladie d'Alzheimer : Alzheimer et bien d'autres maladies sont susceptibles d'être provoqués par des protéines se pliant pour former une structure appelée amyloïdes . Une grande avancée a été faite récemment par le groupe de recherche de David Eisenberg's à UCLA en résolvant la première structure d'une amyloïde. Nous collaborons avec leur groupe de recherche pour utiliser la structure de manière à prédire quelles partie des protéines sont succeptibles de former des amyloïdes, ce qui serait un premier pas vers le blocage de la formation des amyloïdes, et on espère, de la maladie.
- Cancer : Le cancer peut être causé par des mutations des gènes clés qui rompent les processus normaux de contrôle cellulaire. Nous développons des méthodes pour couper l'ADN à des endroits spécifiques dans le génome, et nous ciblerons les endroits impliqués dans le cancer. Après que ces endroits aient été coupés, ils devraient être réparés par la cellule en utilisant une deuxième copie (n'ayant pas muté) du gène et la cellule ne devrait plus être cancéreuse. C'est une méthode très spécifique de thérapie génétique qui, en cas de succès, contournera l'une des principales objections aux méthodes de thérapie génétique; concrètement, les méthodes actuelles insèrent une copie non mutée d'un gène aléatoirement dans le génome, et si le point d'insertion s'avère proche d'un ocnogène, la thérapiegenie soignera une maladie mais en causera une autre. Comme notre méthode ciblera des endroits précis au lien d'endroits aléatoires, elle devrait éviter ce piège.
- Cancer de la Prostate : Le Récepteur Androgène (AR) lie la testostérone et est responsable du développement normal de l'homme. Quand le AR devient hypersensible à la testostérone, le résultat est un cancer de la prostate. Le traitement actuel du cancer de la prostate, appelé "thérapie hormonale", fait baisser le taux de testostérone disponible (parfois par castration). De nombreuses tumeurs malignes sont résistantes à cette thérapie, mais nous appliquons nos méthodes de modélisation des protéines pour trouver des façons différentes d'inhiber l'AR et de traiter le cancer de la prostate. En clair, nous allons essayer de modéliser des protéines qui vont désactiver l'AR en présence de testostérone. Nous faisons cela en concevant des protéines qui vont empêcher l'AR d'entrer dans le noyau de la cellule (là où il fait sa sâle besogne), mais aussi l'empêcher de se lier à l'ADN pour activer des gènes spécifiques aux tumeurs même s'il entre dans le noyau..
Les projets ci-dessus ne fonctionnent pas sur BOINC parce que nous n'avons pas encore un système de file d'attente efficace qui permettrait aux scientifiques de soumettre des travaux simplement, mais attendez-vous à les voir prochainement ! De plus, soyez assurés que les calculs de prédictions de structures qui tournent actuellement sur vos ordinateurs auront un impact direct sur le traitement des maladies. Il est possible de donner une explication en 3 points à propos de cette relation directe entre la prédiction des structures et le traitement des maladies :
1. La prédiction des structures et la conception des protéines sont très liées.
Les améliorations dans la prédiction des structures conduisent à des améliorations dans la conception des protéines, qui permettent de se traduire par la conception de nouveaux enzymes, vaccins, etc... Pour plus d'information sur la modélisation des protéines, vous pouvez consulter les revues que nous avons récemment publiées et qui sont disponibles sur notre page d'accueil ( http://depts.washington.edu/bakerpg )
Schueler-Furman, O., Wang, C., Bradley, P., Misura, K., Baker, D. (2005). Progress in modeling of protein structures and interactions Science 310, 638-642.
2. La prédiction des structures identifie des cibles pour de nouveaux médicamments.
Quand nous prédisons des structures à grande échelle pour les protéines du génome humain , nous en apprenons plus sur les fonctions de beaucoup de ces protéines, ce qui aide à comprendre comment les cellules travaillent et comment les maladies arrivent. Plus directement, nous serons capables d'identifier beaucoup de nouvelles cibles potentielles de médicaments pour lesquelles de petits inhibiteurs de cellules (médicaments) pourront être conçus. Pour remettre cela dans le contexte, l'un des principaux obstacles au développement de nouveaux traitements pour les maladies humaines, tient dans la capacité à identifier de nouvelles cibles de protéines " à soigner". La plupart des nouveaux médicamments actuels interagissent avec les mêmes cibles que les anciens médicamments, donc ces médicamments ne conduisent qu'à de faibles améliorations dans le traitement des maladies. La prédiction des structures nous aide à identifier de nouvelles cibles pour les médicaments et nous aidera donc à trouver des traitements innovants, peut être même révolutionnaires, pour les maladies.
3. La prédiction des structures nous permet d'utiliser une méthode de "conception rationnelle" pour créer de nouveaux médicaments.
Si nous connaissons la structure d'une protéine, nous pouvons déterminer ses sites fonctionnels, et cibler précisément ces sites pour qu'ils soient désactivés par un nouveau médicamment. Le calcul d'une petite molécule (médicament) qui sera liée et inactivera une protéine cible sera similaire en de nombreux points aux calculs de prédiction de structure que nous réalisons ici --c'est en résumé le problème de trouver la structure de moindre énergie d'une protéine ainsi que du système actif du médicamment-- et nous avons récemment développé un nouveau module dans ROSETTA pour effectuer ce travail de liaison. Les résultats sont très prometteurs, et dans un futur proche, vos ordinateurs calculeront probablement les laisons médicamenteuses avec des projets de vaccin et des conceptions de proteines thérapeutiques comme décrits ci-dessus, en plus des calculs de pliage de protéines que vous effectuez en ce moment.
Prédiction et Conception de Structures et d'Interactions Macromoléculaires
L'objectif de notre recherche actuelle est de développer un modèle amélioré pour les interactions intra et inter molléculaires et d'utiliser ce modèle pour prédire et concevoir des structure macromolléculaires et leurs interactions. Les applications de conception et de prédiction, qui peuvent être d'un grand intérêt biologique dans leur propre domaine, fournissent également des tests objectifs et rigoureux qui améliorent le modèle et la compréhension fondamentales.
Nous utilisons un programme informatique appelé Rosetta pour exécuter les calculs et la conception des protéines. Au coeur de Rosetta, il y a des fonctions pour calculer les énergies des interactions entre et à l'intérieur les macromolécules, et des méthodes pour trouver la structure de moindre énergie d'une séquence d'acide aminé (prédiction protéine-structure) ou d'un complexe protéine-protéine, et pour trouver la moindre énergie d'une séquence d'acide aminé pour une protéine ou un complexe protéine-protéine (conception de protéine). Le retour d'information des tests de prédiction et de conception est utilisé continuellement pour améliorer des fonctions potentielles et la recherche d'algorithmes. Le développement d'un programme informatique pour traiter ces divers problèmes a de considérables avantages : d'abord, les différentes applications fournissent des tests complémentaires au modèle physique général (la chimie physique/ physique fondamentale est, bien entendu, la même dans n'importe quel cas); ensuite, de nombreux problèmes d'actualité, comme la conception de structure flexible de protéine et l'assemblage protéine-protéine avec flexibilité de structure, induisent une combinaison de différentes méthodes d'optimisation.
Au cours des dernières années, nous avons utilisé notre méthode de conception par calcul de protéines pour stabiliser de façon remarquable plusieurs petites protéines en modifiant chaque chaîne latérale de leurs séquences, pour revoir la conformation de la chaîne principale de protéines, pour convertir une protéine monomère en dimère par translocation de brins, et pour thermostabiliser une enzyme. Un des moments clés a été de modifier la séquence de repliement de la protéine G, une petite protéine qui contient deux 'bêta-hairpins' (épingles à cheveux) séparées par une 'alpha-hélix' (hélice). Dans la version naturelle de cette protéine, la première épingle est perturbée et la vitesse de formation de la deuxième constitue le facteur limitant la vitesse de pliage. L'ordre des évènements est inversé pour une variante corrigée dans laquelle la première épingle est significativement stabilisée et la deuxième épingle perturbée: la première épingle est formée et la deuxième épingle perturbée vers l'état de transition de repliement. La possibilité de reconcevoir de façon rationnelle les séquences de repliement de protéines montre que notre compréhension des phénomènes réglementant le repliement de protéines a considérablement avancé.

Figure 1: Modélisation de protéines et des interactions protéines-protéines avec une précision haute résolution. Comparaison de modélisation d'une structure structure cristalline (à gauche) une interface représentant une endonuclease avec les nouvelles spécifications des clivages de l'ADN, et (à droite) la nouvelle représentation de la protéine TOP7.
a gauche, Tanja Kortemme. a droite, Gautam Dantas.
Récemment, la création de protéines originales aux structures tri-dimensionnelles arbitrairement choisies a été particulièrement passionnante. Nous avons développé une stratégie générale de calcul pour créer ces structures de protéines qui intègre la flexilibité de chaîne principale à l'optimisation des séquences à l'aide de rotamères. Ceci a pu être accompli en intégrant dans Rosetta les prédictions ab initio de structures de protéines, le raffinement des énergies au niveau atomique, et la conception de séquences. Cette procédure a été utilisée pour concevoir une protéine à 93 chaînes latérales appelée TOP7 avec une séquence et une topologie originales. TOP7 se révéla monomérique et repliée, et la structure cristalline de TOP7 révélée aux rayons X ressemble de façon saisissante (RMSD = 1.2 Angström; voir partie droite de la Figure 1) au modèle calculé. La conception d'un nouveau repli de protéine globulaire et la similarité importante entre la structure cristalline et le modèle calculé ont de vastes implications pour la conception de protéines et pour la prédiction de leurs structures, et ouvrent la voie à l'exploration de vastes domaines de l'univers des protéines pas encore observés dans la nature.
Pour étendre ces méthodes aux interactions protéine-protéine et particulièrement à la reconception de leurs spécificités d'interaction, nous avons choisi le complexe de haute affinité entre la colicine E7 DNase et sa protéine apparentée inhibitrice d'immunité comme système modèle. Nous avons utilisé le système modèle décrit ci-dessus et une modification de notre stratégie de conception par calcul basée sur la recherche de rotamères pour générer de nouvelles paires protéine DNase - protéine inhibitrice supposées interagir étroitement les unes avec les autres mais pas avec des protéines d'origine naturelle. Les complexes de protéines ainsi conçus ont des affinités sub-nanomolaires, sont fonctionnels et spécifiques in vivo, et ont des différences d'affinité entre paires de même origine et paires d'origines différentes de plus d'un ordre de grandeur in vitro. Cette approche devrait pouvoir être appliquée à la conception de paires de protéines interactives possédant des spécificités originales pour représenter et reconcevoir les réseaux d'interaction entre protéines dans les cellules vivantes.
En collaboration avec les équipes de recherche de Barry Stoddard et Ray Monnat (Centre Fred Hutchinson de Recherche Contre le Cancer), nous avons généré une endonucléase artificielle extrêmement spécifique en fusionnant différents domaines d'endonucléases I-DmoI et I-CreI à mécanisme de transfert par transposition ciblée (?) grâce à l'optimisation par calcul d'une nouvelle interface domaine-domaine entre ces deux protéines qui n'interagissent normalement pas entre elles. L'enzyme résultante, E-DRei (pour 'Engineered I-DmoI/I-CreI'), se lie à un long site-cible de chimère ADN à affinité nanomolaire, le divisant à une vitesse équivalente à celle de ses parents naturels (?). Nous sommes actuellement en train d'essayer de générer de nouvelles endonucléases en étendant notre méthodologie de conception aux interfaces protéine - acide nucléique pour reconcevoir l'interface protéine-ADN.
Il a été possible pour ces deux systèmes de déterminer les structures cristallines des complexes conçus grâce aux rayons X. Comme dans le cas de la protéine TOP7, les structures ainsi observées sont très proches des modèles calculés (Figure 1, partie gauche), ce qui valide la précision de notre approche de modélisation de haute résolution.
- Prédiction de la structure de protéines
La représentation du repliement de protéines sur laquelle est basée notre approche de prédiction ab initio de structure tertiaire de protéines est que les interactions locales qui dépendent de la séquence (structure primaire) influencent des segments de la chaîne et définissent plusieurs séries de structures locales distinctes, et que les interactions non-locales définissent les structures tertiaires de plus faible énergie parmi les nombreuses conformations compatibles avec ces influences locales. En appliquant la stratégie suggérée par cette représentation, nous utilisons différents modèles pour traiter les interactions locales et non-locales. Plutôt que d'essayer d'élaborer un modèle physique décrivant les relations entre les séquences et les structures locales, nous nous tournons vers la base de données de protéines et utilisons la distribution de structures locales adoptées par des segments de courtes séquences (moins de 10 résidus de longueur) en structures tri-dimensionnelles connues comme une approximation de la distribution de structures définies par des peptides isolés pour la séquence correspondante.
Les interactions non locales primaires considérées sont mortellement hydrophobes, électrostatiques, liés à l'hydrogène par la chaine principale, et d'un volume exclusif. Les structures qui ont une concordance simultanée avec à la fois la séquence de structure locale influente et les interactions non locales, sont générées en utilisant la recuite simulée pour minimiser l'énergie d'interaction non local dans l'espace défini par les répartitions des structure locales.

Figure 2: Prédictions de structure aveugle de CASP3 et CASP4.
A : A gauche, structure en crital de la transcription du facteur lié à l'ADN 'MarA' ; a droite, notre meilleur modèle moumis dans CASP3. Malgrès beaucoup de détails incorrect, la pliure globale est prédite avec suffisemment d'exactitude pour permettre des apercus à l'intérieur du mode de liaison de l'ADN.
B : A gauche, la structure en cristal du bacteriocine AS-48 ; au milieu, notre meilleur modèle soumis dans CASP4 ; à droite, une protéine apparentée à une autre structurée et fonctionnelle (NK-lysin) identifiée en utilisant ce modèle dans une recherche basée sur la structure de la banque de donnée des protéines (PDB). La similarité structurelle et fonctionnelle n'est pas reconnaissable en utilisant les méthodes de comparaison séquentielles (l'identification entre les deux séquences est seulement de 5 pour cent).
C : A gauche, une structure en cristal du second domaine de MutS ; au milieu, notre meilleur modèle pour ce domaine soumis dans CASP4 ; à droite, une protéine structurellement proche (RuvC) avec une fonction apparentée reconnue en utilisant le modèle d'une recherche basée sur la structure issu de la PDB. La similitude n'était pas reconnus en utilisant les méthodes de comparaison séquentielle ou de reconnaissance de pliure.
Image: Rich Bonneau

Figure 3: La première finalisation d’une résolution de niveau atomique d’une prédiction aveugle de structure ab initio – CASP6 T281. La méthodologie de raffinement haute résolution décrite dans le texte a produit un modèle à1,5 Angström RMSD près de la structure en cristal (à gauche), avec les aspects du paquetage de la chaine latérale originelle (à droite).
Image: Phil Bradley
Un des points forts de CASP6 était la prédiction du premier 'de novo' aveugle qu'a utilisé notre méthodologie d'amélioration haute résolution pour atteindre une exactitude proche de la haute résolution. La séquence relativement courte (76 éléments) nous permet d'appliquer notre méthodologie d'amélioration atome par atome non seulement pour les séquences originelles, mais aussi pour les séquneces de beaucoup homologues. Le centre du groupe d'énergie le plus bas des structures se déplace pour être remarquablement fermé pour les structures natives (1,5 Angström, Figure 3). Le protocole d'amélioration haute résolution réduit le RMSD de 2,2 à 1,5 Angström, et le jeu de chaines latérales dans un comportement assez ressemblant à un l'original dans le centre des protéines (Figure 3, partie droite).
Nous avons étendu la stratégie de prédiction ab initio de structure de Rosetta au problème de l'utilisation de données expérimentales limitées pour de générer des modèles de protéines. Par incorporation de déplacement chimique, d'informations NOE, et plus récemment d'informations dipolaire couplées dans la procédure de génération de structure de Rosetta, nous avons été cappable de générer bien plus de modèles exacts qu'avec la seule prédiction ab initio de structure, ou lors de l'utilisation des mêmes données limitées avec la méthodologie de génération de structure par résonnance magnétique nucléaire (NMR) conventionnelle. c'est développement récent passionnant que la procédure de Rosetta puisse alors exploiter des données NMR non attribuées et de là contourner la difficulté et l'étape fastidieuse de l'affectation du spectre NMR.
La méthode de prediction de structure ab initio de Rosetta, Celle de détermination de structure NMR basée sur Rosetta, et la nouvelle pour la représentation comparative qui utilise l'approche de novo de Rosetta, afin de modéliser une partie d'une structure (Longues boucles primaires) qui ne pouvait pas l'être à partir d'une base précise sur un modèle de structure analogue, ont toutes été implémentées dans un serveur public appelé Robetta
( Robetta). Ce serveur, qui a un constant Backlog d'utilisateurs à travers le monde, était l'un des meilleurs serveurs à la ronde entièrement automatisé de prédiction de structure dans les tests CASP5 et CASP6.
- Prédiction des interactions protéine-protéine
Depuis de nombreuses années nous avons travaillé sur le perfectionnement de la structure protéique, un vrai défi du fait d'un grand nombre de degrés de liberté. Nous avons été intéressés par l'arrimage entre les protéines parce que, en admettant que les deux partenaires ne subissent pas de changement de configuration de façon significative durant l'arimage, l'espace à chercher -les six degrés de liberté de l'axe protéique en plus des degrés de liberté de la chaine latérale- est beaucoup plus petit. Bien qu'important en lui même, ce problème est une bonne marche à gravir vers un plus épineux problème qu'est le perfectionnement de la structure.
Nous avons développé une nouvelle méthode afin de prévoir les complexes protéiques à partir de coordonnées de composants monomères non liés. Cette méthode utilise une recherche à Monte Carlo en basse résolution et corps rigides, suivie par une optimisation simultanée du déplacement de la chaine principale et des conformations des chaines latérales, avec la procedure de minimisation de Monte Carlo et le modèle physique utilisé dans notre travail de prédiction des structures en haute résolution. L'optimisation simultanée des chaines latérales et des degrés de liberté des corps rigides contraste avec la plupart des autre approches actuelles,lesquels modèlilsent d'assemblage protéine-protéine comme un problème de corps rigide de forme identique, avec les chaines latérales tenu fixées. Nous avons récemment amélioré la méthode (RosettaDock?) en déveploppant l'algotihme qui alloue un échantillonnage efficace aux conformations des chaines latérales hors rotamères durant l'assemblage.

Figure 4: Résultats d'assemblage protéine-protéine avec CAPRI (Evaluation judicieuse des interactions prédites). Superposition des structures compexes de protéine prédites (en bleu) et aux rayons X (en rouge et orange). En vert, une chaine latérale pour laquelle la conformation a été correctement prédite pour transformer la formation du complexe. Dans la partie du haut, le complexe entier. Dans la partie du bas, des détails de l'interface. En plus de l'orientation du corps rigide, les conformations de la plupart des chaines latérales sont correctement prédites.
Image: Ora Furman
La puissance RosettaDock a été mis en valeur dans le récent assemblage protéine-protéine aveugle de CAPRI, qui fut trouvé en décembre 2004. Dans CAPRI, les predicteurs ont donné les structures de deux protéines connues pour former un complexe, et pariés qu'ils allaient prédirent la structure de ce complexe. Les prédictions de RosettaDock pour les cibles sans conformation de chaine principale significative furent frappantes, comme montré dans la figure 4. Non seulement les orientation du corps rigide de deux partenaires furent proche de la perfection, mais également presque toute l'interface des chaines latérales fut modélisée trés précisemment. Ces modèles corrects ressorent clairement comme d'énergie moindre que tous les autres modèles que nous avons générés, ce qui suggère que la fonction potentielle est suffisemment précis.
Ces résultats prometteurs suggèrent que la méthode devrait bientôt être utilisable pour générer de modèles d'importants complexes biologiques à partir des structures des composants isolés, et suggérer plus généralement que la modélisation haute résolution de structures et interactions est de portée infèrieure. Un but clair pour notre travail de prédiction de structure monomérique est d'approcher le niveau de précision de ces modèles.
Notre approche courante pour améliorer les fonctions d'énergie implique une combinaison de calculs de chimie quantique sur des modèles simples de composants, de méthodes issues des la mécanique moléculaire traditionelle, et de l'analyse structurelles de proteine. Nous avons utilisé une telle approche pour développer une liaison hydrogène améliorée. Un résultat particulièrement notable est que la dépendance de l'orientation de la liaison hydrogène en chimie quantique des dimères formamide est remarquablement similaire à celle visible dans les liaisons hydrogène de type chaine latérale-chaine latérale des structures des protéines mais différente de celle des champs de force de la mécanique moléculaire courante, laquelle néglige le caractère covalent de la liaison hydrogène. Le retour d'informations provenant des prédictions et des calculs de conception ont insuflé un élan continu et montré la direction pour améliorer la fonction d'énergie; par exemple, des inadéquations dans notre traitement des intéractions protéine-protéine ont amené au développement récemment d'un modèle des rotamères pour les liaisons hydrogènes en solution aqueuse.
- Plans pour le futur
Nos méthodes de prédiction et de représentation ont maintenant abouties un tel niveau qu'elles peuvent être appliqués à d'importants problèmes biologiques. Un point encourageant est qu'après des années de travail de modélisation haute résolution, nous sommes proches des résolutions atomique des prédictions de structures de complexes réalisées par CAPRI (figure 4), les prédictions du CASP6 (figure 3), et très proche du TOP7 (figure 1 , à droite) et l'interface de modélisation protéine-protéine avec structures cristalline par rayon X sont particulièrement encourageants. Ces résultats suggèrent que la modélisantion haute résolution commence à fonctionner.
Dans les prochaines années à venir. Nous avons pour but prinicpal d'améliorer et d'étendre nos méthodes. Nous sommes particulièrement focalisés sur l'amélioration de la précision des prédictions haute résolution de structure (lesquelles sont recquise pour disposer d'un modèle utilisable). Pour en arriver là, nous travaillerons à l'amélioration du modèle physiques sous-jacents et de la méthodologie d'échantillonnage. Nous sommes donc en train de développer les méthodes pour prédire et représenter les spécificités d'interactions ADN-protéines, et d'étendre notre méthode de représentation des protéines pour le traçage des enzymes qui catalysent les réactions chimiques non produites par des protéines naturelles.
Visitez notre site officiel http://www.bakerlab.org pour plus d'informations incluant la liste de nos publications scientifiques. (en anglais)
Les meilleures prévisions réalisées par les membres de l'Alliance Francophone
4 Avril 2006 - 1hz6 _FA_RLX_hom015
Découvreur : [AF>FRANCE>Est>IDF>EDLS] Mephisto94
11 Aout 2007 - CNTRL_01RELAXNATIVE_SAVE_ALL_OUT_-1fkb_-_1801_0
Découvreur : [AF>XTC] Robrouge
20 Octobre 2007 - ccr19_200_BOINC_MFR_ABRELAX_PICKED_1974_0
Découvreur : RvP_LaN
7 Avril 2008 - 1jza__BOINC_ABINITIO_SAVE_ALL_OUT-1jza_-nd__2765_0
Découvreur : sdusart
6 Mai 2008 - t027_1_NMRREF_1_t027_1_id_model_06_2934_0
Découvreur : Big-Buen
18 Aout 2008 - t443__d011a_CASP8_LONGRANGE_JUMP_SAVE_ALL_OUT_BARCODE__4127_0
Découvreur : [AF>DoJ]refresh
15 Septembre 2008 - t466__CASP8_JUMPAB_CROSSBETA_SAVE_ALL_OUT_SELECT_BARCODE_hom001__4216_0
Découvreur : [AF>France>Est>Alsace] Thierry
Représentation de la structure prédite par Mephisto le 4 avril 2006 :
| La plus basse structure énergétique est représentée sur ce graphique en bleu toutes les autres prévisions sont en rouge |
|---|



