


 Au cours des derniers mois, l'équipe a travaillé très dur pour lancer une nouvelle initiative en collaboration avec l'initiative IBM World Community Grid: le projet OpenPandemics: COVID-19.
Au cours des derniers mois, l'équipe a travaillé très dur pour lancer une nouvelle initiative en collaboration avec l'initiative IBM World Community Grid: le projet OpenPandemics: COVID-19.
L'objectif du projet est de cribler virtuellement de très grandes collections de composés chimiques pour identifier les inhibiteurs potentiels du virus SARS-CoV2 qui peuvent fournir un point de départ pour le développement de nouveaux outils thérapeutiques pour COVID-19.
Après avoir fait face à plusieurs défis, nous avons finalement lancé le projet et nous avons déjà recueilli de nombreux résultats qui sont actuellement analysés. Chaque composé que nous identifierons dans nos criblages virtuels sera testé expérimentalement dans des tests in vitro par nos collaborateurs (chez Scripps et ailleurs), et nous espérons évoluer pour devenir des antiviraux COVID-19.
Notre projet est l'un des nombreux efforts actifs qui tentent de trouver de nouveaux médicaments potentiels pour lutter contre cette pandémie. Ce qui est unique dans notre projet, c'est que nous utilisons de nouveaux protocoles d'amarrage moléculaire, ainsi que des approches plus conventionnelles, qui nous permettent de cibler des résidus et des régions spécifiques des protéines virales d'une manière très difficile pour le virus de s'échapper.
Jusqu'à présent, nous avons été très occupés à mettre en place l'infrastructure logicielle, à préparer les données d'entrée à traiter par les volontaires et à nous préparer à recevoir et à analyser l'énorme quantité de données qui va être retournée.
Merci à tous pour votre contribution !
Cet article est un résumé ( attendu depuis longtemps! ) Pour tous les volontaires, couvrant la stratégie de dépistage virtuel et les détails sur les cibles du SRAS-CoV-2 qui ont été considérées jusqu'à présent pour le projet OpenPandemics: COVID-19. Il s'agit d'un travail continu et d'autres structures seront ciblées et nous vous tiendrons au courant des nouvelles structures incluses et des premiers résultats à venir.
Bibliothèques Ligand (Diogo, Matthew)
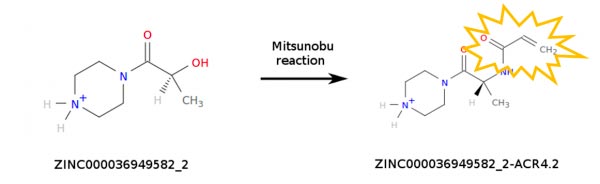 Les calculs effectués par les volontaires du World Community Grid (WCG) simulent des millions de composés chimiques pour aider à identifier ceux qui peuvent interagir avec les protéines du SRAS-CoV-2. Bien qu'il existe plusieurs sources commerciales de composés, nous avons décidé de simuler non seulement des molécules disponibles à l'achat, mais également des molécules pouvant être fabriquées en modifiant celles existantes. En particulier, nous avons envisagé des réactions chimiques qui insèrent une ogive chimique suffisamment réactive pour réagir avec les protéines virales, mais pas trop pour devenir dangereusement toxique. Le but de l'ogive réactive est de former une liaison covalente avec les protéines virales, rendant l'événement de liaison irréversible. Nous n'avons considéré que de simples réactions chimiques pouvant être réalisées par nos collaborateurs en peu de temps. En ce moment, Les volontaires de WCG simulent une bibliothèque de 37 millions de composés contenant l'ogive d'acrylamide qui est spécifique aux résidus de cystéine. De nouveaux composés seront ajoutés à la bibliothèque dans les mois à venir.
Les calculs effectués par les volontaires du World Community Grid (WCG) simulent des millions de composés chimiques pour aider à identifier ceux qui peuvent interagir avec les protéines du SRAS-CoV-2. Bien qu'il existe plusieurs sources commerciales de composés, nous avons décidé de simuler non seulement des molécules disponibles à l'achat, mais également des molécules pouvant être fabriquées en modifiant celles existantes. En particulier, nous avons envisagé des réactions chimiques qui insèrent une ogive chimique suffisamment réactive pour réagir avec les protéines virales, mais pas trop pour devenir dangereusement toxique. Le but de l'ogive réactive est de former une liaison covalente avec les protéines virales, rendant l'événement de liaison irréversible. Nous n'avons considéré que de simples réactions chimiques pouvant être réalisées par nos collaborateurs en peu de temps. En ce moment, Les volontaires de WCG simulent une bibliothèque de 37 millions de composés contenant l'ogive d'acrylamide qui est spécifique aux résidus de cystéine. De nouveaux composés seront ajoutés à la bibliothèque dans les mois à venir.
Structures cibles (Jerome, Giulia, Batuu, Paolo, Martina, Christina)
Dans la phase initiale du projet, nous concentrons nos efforts sur 3 cibles protéiques du SRAS-CoV-2: les protéases de type papaïne (PLpro ou nsp3), la protéase principale (Mpro ou nsp5) et l'endoribonucléase (nsp15 ). Après l'analyse des poches médicamenteuses sur chaque structure, les résidus de cystéine les plus prometteurs, ainsi que les cystéines catalytiques, ont été sélectionnés comme sites covalents potentiels.
1. La première cible que nous considérons est la protéase de type papaïne (PLpro ou nsp3) , qui fait partie de la protéine multi-domaine nsp3. Il est responsable de la maturation des polyprotéines virales et impliqué dans la répression de la réponse immunitaire innée de l'hôte. Dans cette protéine, nous ciblons un résidu spécifique, la cystéine 111, situé dans le site actif et qui joue un rôle clé dans l'activité catalytique. À l'heure actuelle, nous envisageons deux formes différentes de la protéine, l'une de SARS-CoV2 et l'autre de SARS-CoV1 (ID de la banque de données de protéines: 6w9c et 4mm3 ). Pourquoi, demandez-vous? Eh bien, au moment où nous avons commencé ce projet, en mars, peu de structures de SARS-CoV2 étaient disponibles, et une seule (ID PDB: 6w9c) avec la caractéristique clé recherchée: un modèle du résidu cystéine 111 suffisamment accessible pour être ciblé par des liants covalents. Heureusement pour nous, le site actif ne diffère pas beaucoup entre les deux virus SARS-CoV1 et CoV2, ce qui nous permet également d'exploiter une autre structure (PDB id: 4mm3) avec la bonne conformation plus adaptée aux projections virtuelles. Et plus c'est certainement mieux! Depuis, de nombreuses autres structures de nsp3 du SRAS-CoV2 ont été résolues avec d'autres variations structurelles très intéressantes dans le site actif. Et bien sûr, ces conformations seront incorporées dans les prochaines projections !
Cela représente également un défi intéressant car nous pouvons tester une hypothèse clé: pouvons-nous identifier des inhibiteurs viraux à large spectre qui agissent contre plusieurs souches virales? Le savoir est essentiel pour estimer à quel point il sera difficile de lutter contre les futures pandémies.
Pour plus de détails sur PLpro, consultez le billet de blog de nos collaborateurs scientifiques du Coronavirus Structural Task Force en Allemagne, qui fournissent un excellent service à la communauté en améliorant la qualité des structures.
Tous les calculs en cours actuellement visent les différentes conformations de PLpro.
2. La principale protéase (MPro ou nsp5), qui est la cible principale de nombreuses études d'autres laboratoires, est responsable de la maturation de la polyprotéine virale au cours de l'infection. Actuellement, nous envisageons 5 structures pour prendre en compte différentes conformations de la protéine ( ID PDB : 6y84 , 5re9 , 5ren , 6lu7 et 6w63 ). Bien sûr, ils se ressemblent tous de loin, mais quand vous regardez de plus près, certaines petites variations structurelles peuvent faire une énorme différence !
Pour cette protéine, 3 cystéines ont été choisies comme sites de liaison covalente: la cystéine 145 située dans le site actif, la cystéine 156 et la cystéine 300 situées dans des sites de liaison alternatifs, qui n'ont pas encore été explorés. Pour la cystéine 145 principale, les 5 conformations sont utilisées, mais une seule pour la cystéine 156 et la cystéine 300 (ID PDB: 6y84 ). À la fin, nous avons décidé de consacrer plus de temps de calcul à la cystéine active pour laquelle nous savons avec certitude qu'elle peut réagir avec des liants covalents.
Ce sera le prochain objectif sur lequel nous concentrerons nos efforts.
3. L' endoribonucléase spécifique de l'uridylate à ARN nidoviral (NendoU ou nsp15) est une endoribonucléase conservée dans plusieurs familles de virus à ARN, y compris les coronavirus. Bien qu'initialement suspecté de participer directement à la réplication de l'ARN, ses rôles exacts dans le cas du SRAS-CoV et du SARS-CoV-2 sont encore débattus même si plus récemment, il a été suggéré d'interférer avec la réponse immunitaire de l'hôte contre l'ARN double brin intermédiaires, qui sont générés pendant le processus de réplication. Après quelques recherches sur la structure cristalline de NendoU (PDB id: 6vww ), les résidus cystéine 293 (situés dans le site actif) et cystéine 103 ont été choisis comme sites covalents potentiels pour les ogives acrylamide. Une fois les deux premiers objectifs atteints, nous aborderons nsp15.
Concernant la glycoprotéine de pointe externe, qui est la protéine responsable de la fusion et de l'entrée dans les cellules hôtes humaines, nous savons que beaucoup d'entre vous ont demandé pourquoi nous ne ciblons pas actuellement cette protéine. Le principal défi de cette protéine réside dans sa grande flexibilité intrinsèque. Nous savons que la protéine de pointe peut exister principalement dans deux états différents: ouvert ou fermé. Alors pourquoi ne prenons-nous pas simplement ces deux structures? Eh bien, les choses sont plus compliquées car en plus d'être très flexible, cette protéine est également recouverte d'un réseau de sucre complexe qui la protège du système immunitaire de l'hôte et rend difficile la cible directe avec de petites molécules. Nos collaborateurs travaillent à la mise en place de leurs dosages biologiques, nous sommes donc vraiment intéressés à leur fournir des molécules à tester.
Méthode d'amarrage: amarrage réactif (Giulia)
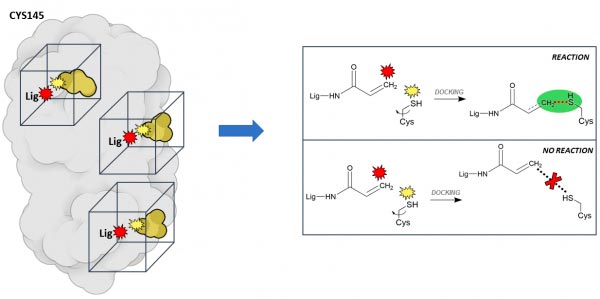 La principale innovation de notre approche réside dans les protocoles d'amarrage que nous utilisons. En plus des protocoles d'amarrage classiques, dans lesquels nous essayons de prédire quelles molécules peuvent se fixer de manière transitoire (c'est-à-dire réversible) à la surface des protéines, nous utilisons un nouveau protocole, appelé "amarrage réactif", pour trouver des liants irréversibles : des molécules qui peuvent se fixer de manière plus serrée et irréversible. L'un des avantages de cette classe de molécules est qu'une fois qu'elles s'accrochent à leur cible, elles la désactivent jusqu'à ce que la protéine soit dégradée, ce qui est clairement une excellente caractéristique à avoir dans un médicament ! et si elles sont bien conçues, elles peuvent être très sélectives. Par exemple, la pénicilline, probablement l'antibiotique le plus connu, fonctionne exactement de cette manière, en réagissant spécifiquement avec les protéines de la paroi des bactéries sans interagir avec les protéines humaines. Le "reactive docking" est un protocole que nous avons développé dans notre laboratoire qui nous permet de simuler la réaction entre le ligand et un résidu covalent spécifique de la protéine. Essentiellement, chaque fois que les atomes "réactifs" du ligand et du résidu se trouvent à une distance de liaison, nous supposons que la réaction est susceptible de se produire. D'un point de vue technique, cela représente un avantage important car nous espérons que cela réduira considérablement les taux de faux positifs (c'est-à-dire les molécules dont on a prédit à tort qu'elles seraient actives), puisque nous pouvons filtrer tous ceux qui n'ont pas réagi. À ce jour, c'est la seule méthode d'arrimage qui permet de réaliser de telles expériences de criblage virtuel sur de très grandes bibliothèques de composés.
La principale innovation de notre approche réside dans les protocoles d'amarrage que nous utilisons. En plus des protocoles d'amarrage classiques, dans lesquels nous essayons de prédire quelles molécules peuvent se fixer de manière transitoire (c'est-à-dire réversible) à la surface des protéines, nous utilisons un nouveau protocole, appelé "amarrage réactif", pour trouver des liants irréversibles : des molécules qui peuvent se fixer de manière plus serrée et irréversible. L'un des avantages de cette classe de molécules est qu'une fois qu'elles s'accrochent à leur cible, elles la désactivent jusqu'à ce que la protéine soit dégradée, ce qui est clairement une excellente caractéristique à avoir dans un médicament ! et si elles sont bien conçues, elles peuvent être très sélectives. Par exemple, la pénicilline, probablement l'antibiotique le plus connu, fonctionne exactement de cette manière, en réagissant spécifiquement avec les protéines de la paroi des bactéries sans interagir avec les protéines humaines. Le "reactive docking" est un protocole que nous avons développé dans notre laboratoire qui nous permet de simuler la réaction entre le ligand et un résidu covalent spécifique de la protéine. Essentiellement, chaque fois que les atomes "réactifs" du ligand et du résidu se trouvent à une distance de liaison, nous supposons que la réaction est susceptible de se produire. D'un point de vue technique, cela représente un avantage important car nous espérons que cela réduira considérablement les taux de faux positifs (c'est-à-dire les molécules dont on a prédit à tort qu'elles seraient actives), puisque nous pouvons filtrer tous ceux qui n'ont pas réagi. À ce jour, c'est la seule méthode d'arrimage qui permet de réaliser de telles expériences de criblage virtuel sur de très grandes bibliothèques de composés.
Architecture de données (Andreas)
Avant que les premiers résultats ne commencent à arriver, nous nous sommes préparés à pouvoir recevoir tous les résultats, les stocker et les rendre disponibles pour le filtrage et l'analyse. Chacune de vos exécutions crée environ 300 Ko de données dans deux fichiers de résultats - l'un avec l'extension DLG et l'autre un fichier XML simple pour une indexation plus facile. L'extension de fichier DLG est le fichier de résultat principal de notre programme (AutoDock4.2) stockant les coordonnées et les énergies des ligands candidats ancrés ( D ocking L o G ).
Alors que 300 Ko par exécution en moyenne peuvent ne pas sembler beaucoup, lorsque vous le multipliez par le nombre d'exécutions que WCG envoie par paquet d'entrée (environ 10000 exécutions), il en ressortira en fait à environ 3 Go. Nous avons commencé à préparer plus de 10 000 colis et espérons aller bien au-delà à terme. Avec la compression (bon vieux Gzip!), Nous sommes en mesure d'abaisser un seul paquet à environ 400 Mo. Sur la base des premières estimations du nombre d'exécutions, nous nous retrouverions avec plusieurs dizaines de TB de données ainsi créées.
Afin de réduire l'utilisation du disque pour stocker une si grande quantité de données, nous avons conçu une méthode pour ramener un seul paquet à environ 85 Mo, soit environ 20% de sa taille compressée, en utilisant la description interne des solutions d'accueil utilisées dans notre logiciel. Typiquement, dans les dockings, il y a une cible fixe (le soi-disant récepteur, c'est-à-dire une protéine donnée du COVID-19) et une molécule flexible à ancrer (le soi-disant ligand, c'est-à-dire un médicament potentiel). Le ligand est déplacé, tourné et sa géométrie est déterminée par des rotations de liaison entre ses atomes. Nous appelons l'ensemble de paramètres qui décrit les propriétés susmentionnées «génome» (puisque nous utilisons un algorithme génétique pour exécuter nos recherches… c'est pour un autre poste…), et comme dans la vie réelle, un génome de ligand peut être utilisé pour le décrire complètement. En stockant uniquement les «informations génétiques» du ligand, nous avons pu réduire la taille des données à environ 20% de leur taille compressée à l'origine. Ces génomes «séchés» réduisent considérablement la quantité de données à transférer et à stocker dans notre base de données.
Alors qu'il vous faut jusqu'à 2,8 années CPU pour trouver le résultat optimal du génome pour un seul paquet, il ne nous faut qu'environ 30 minutes pour régénérer les coordonnées et les énergies d'origine (puisque nous connaissons déjà la solution). Nous disposons de deux serveurs dédiés pour recevoir et stocker les données, et effectuer les opérations de traitement.
Alors, qu'arrive-t-il aux données pendant le traitement ? Il y a deux choses fondamentales qui se produisent dès que nous recevons un fichier de résultats: premièrement, les génomes «séchés» dans les résultats sont «réhydratés», ce qui signifie que les coordonnées de toutes les poses pour chaque course sont calculées dans l'espace tridimensionnel; puis chaque pose est analysée individuellement.
Un script d'analyse recueille les informations sur le type d'interactions (par exemple, polaires / non-interactions, électrostatiques, etc.) et les résidus cibles avec lesquels chaque pose interagit (c'est-à-dire la cystéine 111). Il vérifie également si la molécule a réagi ou non (selon les paramètres d'amarrage réactif). Ces informations sont collectées de manière très efficace et stockées dans notre base de données. Il s'agit d'une étape essentielle dans la phase de traitement car elle nous permet d'effectuer des requêtes très complexes qui nous permettent à terme de rechercher les quelques centaines «d'aiguilles» prometteuses dans une botte de foin contenant des milliards de poses en quelques secondes. Les composés les plus prometteurs seront inspectés visuellement un par un et sélectionnés pour passer à la phase de test. Merci à tout votre soutien, au moment de la rédaction de la base de données en contient environ 3.
traduction de l'article du ForliLab : https://forlilab.org/opn-update-7-20/?linkId=94724376