


Récapitulatif
Dans cette mise à jour complète, l'équipe Mapping Cancer Markers explique comment elle détermine quels gènes et signatures de gènes sont les plus prometteurs pour le diagnostic du cancer du poumon. Ils introduisent également le prochain type de cancer - le sarcome - qui sera bientôt ajouté au projet.![]()
Le projet Mapping Cancer Markers (MCM) continue de traiter les unités de travail pour l'ensemble de données sur le cancer de l'ovaire. À mesure que nous accumulons ces résultats, nous continuons d'analyser les résultats MCM de l'ensemble de données sur le cancer du poumon. Dans cette mise à jour, nous discutons des résultats préliminaires de cette analyse. De plus, nous introduisons l'ensemble de données sur le sarcome qui sera notre objectif dans la prochaine étape.
Modèles de biomarqueurs de la famille des gènes dans le cancer du poumon
Dans le cancer et la biologie humaine en général, plusieurs groupes de biomarqueurs (gènes, protéines, microARN, etc.) peuvent avoir des profils d'activité similaires et donc une utilité clinique, facilitant le diagnostic, le pronostic ou la prédiction des résultats du traitement. Pour chaque sous-type de cancer, on pourrait trouver un grand nombre de ces groupes de biomarqueurs, chacun ayant un pouvoir prédictif similaire; pourtant, les méthodes statistiques et basées sur l'IA actuelles n'en identifient qu'une parmi un ensemble de données donné.
Nous avons deux objectifs principaux dans MCM: 1) trouver de bons groupes de biomarqueurs pour les cancers que nous étudions, et 2) identifier comment et pourquoi ces biomarqueurs forment des groupes utiles, afin que nous puissions construire une approche heuristique qui trouvera de tels groupes pour tout maladie sans avoir besoin de mois de calcul sur World Community Grid. Le premier objectif nous donnera non seulement des informations qui, après validation, peuvent être utiles dans la pratique clinique, mais surtout, elles généreront des données que nous utiliserons pour valider nos heuristiques.
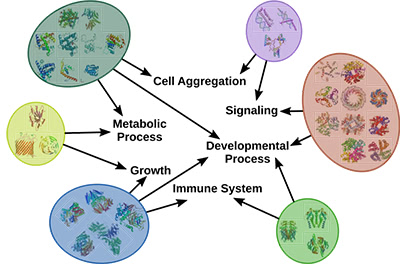
Illustration 1: Groupe de protéines par interactions similaires et fonctions biologiques similaires.
De multiples groupes de biomarqueurs existent principalement en raison de la redondance et du câblage complexe du système biologique. Par exemple, le réseau d'interaction protéine-protéine humaine hautement interconnecté nous permet de voir comment les protéines individuelles remplissent diverses fonctions moléculaires et contribuent ensemble à un processus biologique spécifique, comme le montre l'illustration 1 ci-dessus. Beaucoup de ces interactions changent entre l'état de santé et l'état de maladie, qui à son tour affecte les fonctions de ces protéines. À travers ces analyses, nous visons à construire des modèles de ces processus qui pourraient à leur tour être utilisés pour concevoir de nouvelles approches thérapeutiques.
Deux groupes spécifiques de biomarqueurs peuvent sembler différents l'un de l'autre, tout en étant équivalents car les protéines remplissent des fonctions moléculaires similaires. Cependant, l'utilisation de ces groupes de biomarqueurs pour la stratification des patients peut ne pas être simple. Les groupes de biomarqueurs ne sont souvent pas validés dans de nouvelles cohortes de patients ou lorsqu'ils sont mesurés par différents tests biologiques, et il y a des milliers de combinaisons possibles à considérer. Certains groupes de biomarqueurs peuvent avoir tous les réactifs disponibles tandis que d'autres doivent être développés (ou être plus chers); ils peuvent également avoir une robustesse, une sensibilité et une précision différentes, affectant leur potentiel en tant que biomarqueurs cliniquement utiles.
À l'heure actuelle, il n'y a pas d'approche efficace pour trouver tous les bons groupes de biomarqueurs nécessaires pour atteindre l'objectif défini, comme prédire avec précision le risque du patient ou la réponse au traitement.
Le premier objectif du projet Mapping Cancer Markers est de mieux comprendre les «règles» de pourquoi et comment les protéines interagissent et peuvent être combinées pour former un groupe de biomarqueurs, ce qui est essentiel pour comprendre leur rôle et leur applicabilité. Par conséquent, nous utilisons la ressource informatique unique de World Community Grid pour sonder systématiquement le paysage de groupes de biomarqueurs utiles pour de multiples cancers et objectifs (diagnostic et pronostic). Ainsi, nous avons établi une référence pour l'identification et la validation des biomarqueurs des gènes du cancer. Simultanément, nous appliquons des méthodes d'apprentissage non supervisées telles que le regroupement hiérarchique à des protéines qui se regroupent par pouvoir prédictif et fonction biologique.
La combinaison de ce clustering et des modèles de World Community Grid nous permet d'identifier des clusters de gènes généralisés qui fournissent des informations plus approfondies sur le fond moléculaire des cancers et donnent naissance à des groupes plus fiables de biomarqueurs génétiques pour la détection et le pronostic du cancer.
Actuellement, nous nous concentrons sur les résultats de la première phase de l'ensemble de données sur le cancer du poumon, qui s'est concentré sur une exploration systématique de tout l'espace des groupes potentiels de biomarqueurs de longueur fixe.
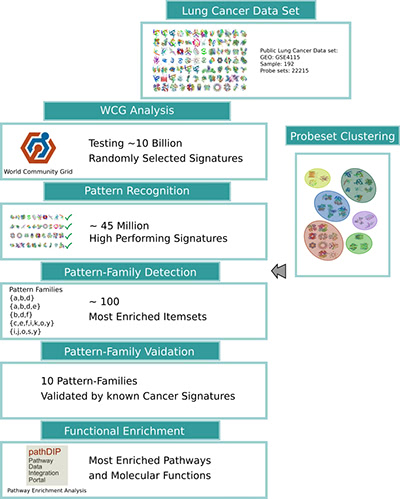
Illustration 2: Workflow de la recherche MCM-gène-pattern-family. Les résultats de l'analyse du World Community Grid combinés au regroupement non supervisé des gènes identifient un ensemble de familles de patrons de gènes, généralisant les groupes de biomarqueurs. Enfin, les résultats sont évalués à l'aide de biomarqueurs du cancer connus et à l'aide d'annotations fonctionnelles, telles que les voies de signalisation, la fonction et les processus d'ontologie génétique.
Comme illustré ci-dessus dans l'illustration 2, World Community Grid a calculé environ 10 milliards de groupes de biomarqueurs sélectionnés au hasard, pour nous aider à comprendre la distribution des tailles de groupe et des combinaisons de biomarqueurs qui fonctionnent bien, que nous utiliserons à notre tour pour valider les approches heuristiques. L'analyse a montré qu'environ 45 millions de groupes de biomarqueurs avaient un pouvoir prédictif élevé et dépassaient le seuil de qualité. Cette évaluation nous donne une image détaillée et systématique des gènes et groupes de gènes porteurs des informations les plus précieuses pour le diagnostic du cancer du poumon. L'ajout de données sur les voies et les réseaux d'interaction des protéines nous permet de mieux interpréter et comprendre comment et pourquoi ces groupes de biomarqueurs fonctionnent bien, et quels processus et fonctions ces protéines transportent.
Simultanément, nous avons utilisé les données décrites sur le cancer du poumon pour découvrir des groupes de gènes similaires. Nous supposons que ces gènes ou les protéines codées remplissent des fonctions biologiques similaires ou sont impliqués dans les mêmes processus moléculaires.
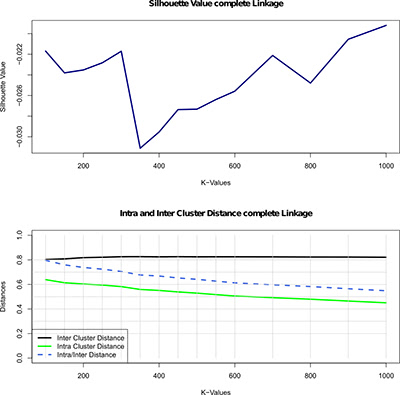
Illustration 3: Évaluation du regroupement hiérarchique des données sur le cancer du poumon, en utilisant le paramètre de liaison complet, pour différents nombres de groupes indiqués par les valeurs K (100 à 1000). Le premier graphique montre la valeur de la silhouette - une métrique de qualité dans ce regroupement, c'est-à-dire la mesure de la relation entre chaque objet et son cluster par rapport aux autres clusters. Le deuxième graphique illustre la distance inter et intra grappe et le rapport de la distance intra / inter grappe.
Pour trouver les algorithmes de clustering appropriés et le bon nombre de groupes de gènes (clusters), nous utilisons différentes mesures pour évaluer la qualité de chaque clustering individuel. Par exemple, l'illustration 3 (ci-dessus) montre les résultats de l'évaluation du clustering hiérarchique pour différents nombres de clusters. Pour évaluer la qualité du clustering, nous avons utilisé la valeur de la silhouette (méthode pour évaluer la cohérence au sein des clusters de données, c'est-à-dire la mesure de la relation entre chaque objet et son propre cluster par rapport aux autres clusters). Une valeur de silhouette élevée indique une bonne configuration de regroupement, et la figure montre une forte augmentation de la valeur de silhouette à 700 groupes de gènes. Comme cela indique une augmentation significative de la qualité, nous sélectionnons ensuite ce regroupement pour une analyse plus approfondie.
Toutes les combinaisons de fonctions biologiques ou leur absence n'entraîneront pas le développement d'un cancer et ne seront pas importantes sur le plan biologique. Dans l'étape suivante, nous appliquons une recherche statistique pour rechercher les combinaisons de grappes les plus courantes parmi les biomarqueurs bien préformés, et donc aboutir à des groupes de gènes ou des familles de motifs. Étant donné que certaines familles de modèles de gènes sont susceptibles de se produire même au hasard, nous utilisons une analyse d'enrichissement pour nous assurer que la sélection ne contient que des familles qui se produisent beaucoup plus souvent que le hasard.
Dans l'étape suivante, nous avons validé les familles de modèles génétiques généralisées sélectionnées en utilisant un ensemble indépendant de 28 ensembles de données sur le cancer du poumon. Chacune de ces études rapporte un ou plusieurs groupes de biomarqueurs de gènes régulés à la hausse ou à la baisse qui sont indicatifs du cancer du poumon.

Illustration 4: Voici une sélection de familles de modèles hautement performantes et la façon dont elles sont prises en charge par 28 signatures de gènes publiées précédemment. Chaque cercle de la figure indique la force du soutien: la taille du cercle représente le nombre de grappes de la famille qui se trouvaient beaucoup plus souvent dans la signature de cette étude. La couleur du cercle indique la signification moyenne calculée pour tous les groupes de la famille de motifs.
L'illustration 4 présente une sélection des familles de modèles les plus répandues et les études qui les soutiennent. Chaque cercle de la figure indique la force du support: la taille du cercle représente le nombre de grappes de la famille qui se trouvent significativement plus souvent dans le biomarqueur de cette étude. La couleur du cercle indique la signification moyenne calculée pour tous les groupes de la famille de motifs.
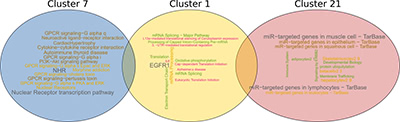
Illustration 5: L'une des familles de patrons de gènes les plus fréquentes est une combinaison des clusters 1, 7 et 21. Nous avons annoté chaque cluster avec des voies à l'aide de pathDIP et l'avons visualisé à l'aide de nuages de mots (plus le mot / phrase est grand, plus le plus souvent ça arrive).
Enfin, nous avons annoté les familles de patrons de gènes les plus efficaces et leurs grappes de gènes avec les fonctions moléculaires et les voies dans lesquelles les gènes ou les protéines correspondantes sont impliquées. L'illustration 5 montre un exemple d'une telle famille de patrons de gènes qui comprend des grappes de gènes 7 , 1 et 21.
La visualisation du nuage de mots indique que le cluster 7 est impliqué dans des voies liées aux GPCR (récepteurs couplés aux protéines G) et aux NHR (récepteurs des hormones nucléaires). En revanche, les gènes du cluster 1 sont hautement enrichis en EGFR1 (récepteur du facteur de croissance épidermique) ainsi que des voies de régulation traductionnelle. Les mutations affectant l'expression d'EGFR1, une protéine transmembranaire, se sont révélées entraîner différents types de cancer, et en particulier le cancer du poumon (comme nous l'avons montré précédemment, par exemple (Petschnigg et al., J Mol Biol 2017; Petschnigg et al. , Nat Méthodes2014)). Les aberrations augmentent l'activité kinase d'EGFR1, conduisant à une hyperactivation des voies de signalisation pro-survie en aval et à une division cellulaire non contrôlée ultérieure. La découverte d'EGFR1 a initié le développement d'approches thérapeutiques contre divers types de cancer, dont le cancer du poumon. Le troisième groupe de gènes sont des cibles communes des microARN. Le groupe 21 indique une forte implication avec les microARN, comme nous et d'autres avons déjà montré (Tokar et al., Oncotarget 2018; Becker-Santos et al., J Pathology , 2016; Cinegaglia et al., Oncotarget 2016).
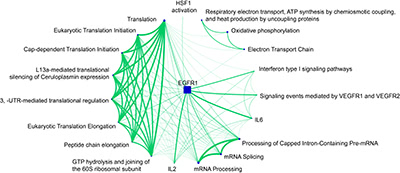
Illustration 6: Évaluation des voies enrichies pour le cluster 1. Ici, nous avons utilisé notre portail d'accès à l'analyse de l'enrichissement des voies pathDIP (Rahmati et al., NAR 2017). Le réseau a été généré avec notre outil de visualisation et d'analyse de réseau NAViGaTOR 3 ( http://ophid.utoronto.ca/navigator ).
L'illustration finale évalue les 20 voies les plus enrichies pour le cluster 1. La taille des nœuds de voie correspond au nombre de gènes impliqués, et la largeur des bords correspond au nombre de gènes de chevauchement entre les voies. On peut voir que toutes les voies impliquées dans la traduction se chevauchent fortement. Les voies liées à l'ARNm forment un autre composant hautement connecté dans le graphique. La voie EGFR1 chevauche fortement la plupart des autres voies, indiquant que les gènes qui sont affectés par ces voies sont impliqués dans un mécanisme moléculaire similaire.
Sarcome
Après les cancers du poumon et de l'ovaire, nous nous concentrerons ensuite sur le sarcome. Les sarcomes sont un groupe hétérogène de tumeurs malignes qui sont relativement rares. Ils sont généralement classés en fonction de la morphologie et du type de tissus conjonctifs dans lesquels ils surviennent, notamment la graisse, les muscles, les vaisseaux sanguins, les tissus cutanés profonds, les nerfs, les os et le cartilage, ce qui représente moins de 10% de toutes les tumeurs malignes (Jain 2010). Les sarcomes peuvent survenir n'importe où dans le corps humain, de la tête aux pieds, peuvent se développer chez des patients de tout âge, y compris les enfants, et varient souvent en agressivité, même au sein d'un même sous-type d'organe ou de tissu (Honore 2015). Cela suggère qu'une description histologique par type d'organe et de tissu n'est pas suffisante pour catégoriser la maladie et n'aide pas non plus à sélectionner le traitement le plus optimal.
Le diagnostic des sarcomes pose un dilemme particulier, non seulement en raison de leur rareté, mais aussi en raison de leur diversité, avec plus de 70 sous-types histologiques, et de notre compréhension insuffisante des caractéristiques moléculaires de ces sous-types (Jain 2010).
Par conséquent, des recherches récentes se sont concentrées sur les classifications moléculaires des sarcomes basées sur des altérations génétiques, telles que les gènes de fusion ou les mutations oncogènes. Bien que la recherche ait permis de réaliser des développements majeurs dans le contrôle local / la récupération des membres, le taux de survie des sarcomes des tissus mous (STS) «à haut risque» ne s'est pas amélioré de manière significative, en particulier chez les patients atteints d'un grand sarcome profond et de haut grade (stade III) Kane III 2018).
Pour ces raisons, dans la prochaine phase de l'analyse de World Community Grid, nous nous concentrerons sur l'évaluation du contexte génomique du sarcome. Nous utiliserons différentes informations et technologies de séquençage pour acquérir une connaissance plus large entre les différents niveaux d'aberrations génétiques et les implications réglementaires. Nous fournirons une description plus détaillée des données et des incitations dans la prochaine mise à jour.
- Petschnigg J, Kotlyar M, Blair L, Jurisica I, Stagljar I et Ketteler R, Identification systématique des partenaires d'interaction EGFR oncogènes, J Mol Biol , 429 (2): 280-294, 2017.
- Petschnigg, J., Groisman, B., Kotlyar, M., Taipale, M., Zheng, Y., Kurat, C., Sayad, A., Sierra, J., Mattiazzi Usaj, M., Snider, J. , Nachman, A., Krykbaeva, I., Tsao, MS, Moffat, J., Pawson, T., Lindquist, S., Jurisica, I., Stagljar, I.Mammalian Membrane Two-Hybrid assay (MaMTH): a nouvel outil à deux hybrides à ubiquitine divisée pour l'étude fonctionnelle des voies de signalisation dans les cellules humaines; Nat Methods , 11 (5): 585-92, 2014.
- Rahmati, S. , Abovsky, M., Pastrello, C. , Jurisica, I. pathDIP: une ressource annotée pour les associations connues et prédites de gènes-voies humaines et l'analyse d'enrichissement de voies. Nucl Acids Res , 45 (D1): D419-D426, 2017.
- Kane, John M. et al. "Corrélation des modèles d'expression de biomarqueurs à sarcome des tissus mous à haut risque avec le résultat après la chimioradiothérapie néoadjuvante." Sarcome 2018 (2018).
- Jain, Shilpa et al. "Classification moléculaire des sarcomes des tissus mous et ses applications cliniques." Revue internationale de pathologie clinique et expérimentale 3.4 (2010): 416.
- Honoré, C., et al. "Sarcome des tissus mous en France en 2015: épidémiologie, classification et organisation des soins cliniques." Journal de chirurgie viscérale 152.4 (2015): 223-230.
- Tokar T , Pastrello C , Ramnarine VR, Zhu CQ, Craddock KJ, Pikor L, Vucic EA, Vary S, Shepherd FA, Tsao MS, Lam WL , Jurisica Les microARN exprimés différentiellement dans l'adénocarcinome pulmonaire inversent les effets des aberrations du nombre de copies des gènes pronostiques. Oncotarget . 9 (10): 9137-9155, 2018
- Becker-Santos, DD, Thu, KL, anglais, JC, Pikor, LA, Chari, R., Lonergan, KM, Martinez, VD, Zhang, M., Vucic, EA, Luk, MTY, Carraro, A., Korbelik , J., Piga, D., Lhomme, NM, Tsay, MJ , Yee, J., MacAulay, CE, Lockwood, WW, Robinson, WP, Jurisica, I. , Lam, WL, Facteur de transcription développemental NFIB est un putatif cible des miARN oncofœtaux et est associée à l'agressivité tumorale dans l'adénocarcinome pulmonaire, J Pathology , 240 (2): 161-72, 2016.
- Cinegaglia , NC , Andrade, SCS, Tokar, T. , Pinheiro, M., Severino, FE, Oliveira, RA, Hasimoto, EN, Cataneo, DC, Cataneo, AJM, Defaveri, J., Souza, CP, Marques, MMC , Carvalho, RF, Coutinho, LL, Gross, JL, Rogatto, SR, Lam, WL, Jurisica, I. , Reis, PP L'analyse intégrative du transcriptome identifie les réseaux de facteurs de transcription microARN dérégulés dans le poumon, l'adénocarcinome, Oncotarget , 7 (20) : 28920-34, 2016.
Autres actualités
Nous avons obtenu un financement important du gouvernement de l'Ontario pour notre recherche: la plateforme de biologie de la signalisation de nouvelle génération . L'objectif principal du projet est de développer une nouvelle plate-forme analytique intégrée et un flux de travail pour la médecine de précision. Ce projet créera une ressource accessible à l'échelle internationale qui unifie différents types de données biologiques, y compris les informations personnelles sur la santé, libérant ainsi tout son potentiel et la rendant plus utilisable pour la recherche dans le continuum de la santé: des gènes et des protéines aux voies, aux médicaments et aux humains.
Nous avons également publié des articles décrivant plusieurs outils, portails et applications avec nos collaborateurs. Ci-dessous, nous listons les personnes les plus directement ou indirectement liées au travail sur World Community Grid:
- Wong, S. , Pastrello, C., Kotlyar, M., Faloutsos, C., Jurisica, je . SDREGION: Repérage rapide des communautés changeantes dans les réseaux biologiques. ACM KDD Proceedings , 2018. Sous presse. BMC Cancer, 18 (1): 408, 2018.
- Kotlyar, M., Pastrello, C., Rossos, A., Jurisica, I. Bases de données d'interaction protéine-protéine. Eds. Cannataro, M. et al. Encyclopédie de bioinformatique et de biologie computationnelle , 81 , Elsevier. Dans la presse. doi.org/10.1016/B978-0-12-811414-8.20495-1
- Rahmati, S., Pastrello, C., Rossos, A., Jurisica, je . Deux décennies de bases de données sur les voies biologiques: résultats et défis, Eds. Cannataro, M. et al. Encyclopédie de bioinformatique et de biologie computationnelle , 81 , Elsevier. Dans la presse.
- Hauschild, AC , Pastrello, C., Rossos, A., Jurisica, je . Visualization of Biomedical Networks, Eds. Cannataro, M. et al. Encyclopédie de bioinformatique et de biologie computationnelle , 81 , Elsevier. Dans la presse.
- Sivade Dumousseau M, Alonso-López D, Ammari M, Bradley G, Campbell NH, Ceol A, Cesareni G, Combe C, De Las Rivas J, Del-Toro N, Heimbach J, Hermjakob H, Jurisica I , Koch M, Licata L, Lovering RC, Lynn DJ, Meldal BHM, Micklem G, Panni S, Porras P, Ricard-Blum S, Roechert B, Salwinski L, Shrivastava A, Sullivan J, Thierry-Mieg N, Yehudi Y, Van Roey K, Orchard S. Nouveaux cas d'utilisation englobants - niveau 3.0 du format HUPO-PSI pour les interactions moléculaires. BMC Bioinformatics, 19 (1): 134, 2018.
- Minatel BC, Martinez VD, Ng KW, Sage AP, Tokar T , Marshall EA, Anderson C, Enfield KSS, Stewart GL, Reis PP, Jurisica I , Lam WL., Découverte à grande échelle de microARN auparavant non détectés spécifiques au foie humain. Hum Genomics, 12 (1): 16, 2018.
- Tokar T , Pastrello C , Ramnarine VR, Zhu CQ, Craddock KJ, Pikor L, Vucic EA, Vary S, Shepherd FA, Tsao MS, Lam WL , Jurisica, I.Les microARN exprimés différentiellement dans les adénocarcinomes pulmonaires inversent les effets des aberrations du nombre de copies de gènes pronostiques. Oncotarget . 9 (10): 9137-9155, 2018.
- Paulitti A, Corallo D, Andreuzzi E, Bizzotto D, Marastoni S, Pellicani R, Tarticchio G, Pastrello C , Jurisica I , Ligresti G, Bucciotti F, Doliana R, Colladel R, Braghetta P, Di Silvestre A, Bressan G, Colombatti A, Bonaldo P, Mongiat M. L'ablation de la protéine EMILIN2 matricellulaire ca 1 utilise une vascularisation défectueuse due à une production altérée d'IL-8 dépendante de l'EGFR, Oncogene , 27 février. Doi: 10.1038 / s41388-017-0107-x. [Epub avant l'impression] 2018.
- Tokar, T. , Pastrello, C., Rossos, A., Abovsky, M., Hauschild, AC, Tsay, M., Lu, R., Jurisica. I. mirDIP 4,1 - Basedonnées intégrative de prévisions decible de microARN humains, Nucl Acids Res, D1 (46): D360-D370, 2018.
- Kotlyar M., Pastrello, C., Rossos, A., Jurisica, I. , Prediction of protein-protein interactions, Current Protocols in Bioinf , 60 , 8.2.1–8.2.14., 2017.
- Singh, M., Venugopal, C., Tokar, T., Brown, KB, McFarlane, N., Bakhshinyan, D., Vijayakumar, T., Manoranjan, B., Mahendram, S., Vora, P., Qazi , M., Dhillon, M., Tong, A., Durrer, K., Murty, N., Hallet, R., Hassell, JA, Kaplan, D., Jurisica, I. , Cutz, JC., Moffat, J., Singh, DK, RNAi screen identifie les régulateurs essentiels des cellules initiatrices de métastases cérébrales humaines, Acta Neuropathologica , 134 (6): 923-940, 2017.
Je vous remercie
Ce travail ne serait pas possible sans la participation des membres du World Community Grid. Merci d'avoir généreusement contribué aux cycles CPU et de l'intérêt que vous portez à ce projet et aux autres projets de World Community Grid.
| Par: dr. Igor Jurisica |
| Institut de recherche Krembil, University Health Network, Toronto 8 août 2018 |
Traduction de la page du site WCG : https://www.worldcommunitygrid.org/about_us/viewNewsArticle.do?articleId=571