


|
|

Etats alternatifs des protéines mis en évidence par la cartographie détaillée de l'énergie de surface :
Quelles conformations les molécules de protéines prennent en solution ? La cristallographie fournit une représentation haute résolution de la structure des protéines dans un environnement cristallin, tandis que la résonance magnétique nucléaire (RMN) donne une structure en solution, mais en utilisant moins de données. Les structures obtenues par résonance magnétique nucléaire sont plus hétérogènes, mais est-ce dû au fait que les contacts cristallins sont absents ou est-ce en raison du nombre de données moins importantes ? Nous apportons ici un aperçu inattendu de cette question obtenue par l'analyse détaillée des énergies de surface protéiques générées à grande échelle, sur un échantillonnage natif de l'espace conformationnel avec Rosetta@home pour 111 domaines de protéines. En l'absence de partenaire ou de ligand étroitement lié, la plus faible énergie des modèles Rosetta est presque toujours inférieure à 2.5 Ångström Cα RMSD par rapport à la structure expérimentale. Ce résultat démontre que la précision de prédiction de la structure des protéines globulaires est principalement limitée par la capacité d'échantillonnage proche de la structure native.
Bien que les modèles de plus basse énergie soient similaires à la structure déposée dans la banque de données des protéines du Research Collaboratory for Structural Bioinformatics (Protein Data Bank ou PDB), ils ne sont pas identiques. Les plus grandes divergences sont le plus souvent localisées au niveau des régions impliquées dans les interactions avec les ligands, les structures quaternaires, ou des contacts cristallins. Pour des protéines liées à un ligand, les modèles de basse énergie peuvent ressembler à des structures apo, et pour les protéines oligomériques, à l'ensemble des molécules monomériques. Les écarts entre les modèles à basse énergie et les structures cristallines disparaissent en grande partie lorsque les surfaces sont calculées dans le contexte du réseau cristallin ou multimérique. Les résultats des formes de basse énergie calculés à partir de la surface englobant la structure cristalline, mais surtout avec la structure RMN apportant une variabilité dans les boucles, peuvent dans certains cas, se rapprocher de l'état natif de ces ensembles de protéines et ce bien mieux que les structures cristallines ou de RMN. Cela peut suggérer des hypothèses testables expérimentalement par rapport à des états alternatifs et à l'hétérogénéité structurale au niveau fonctionnel.
|
|
|
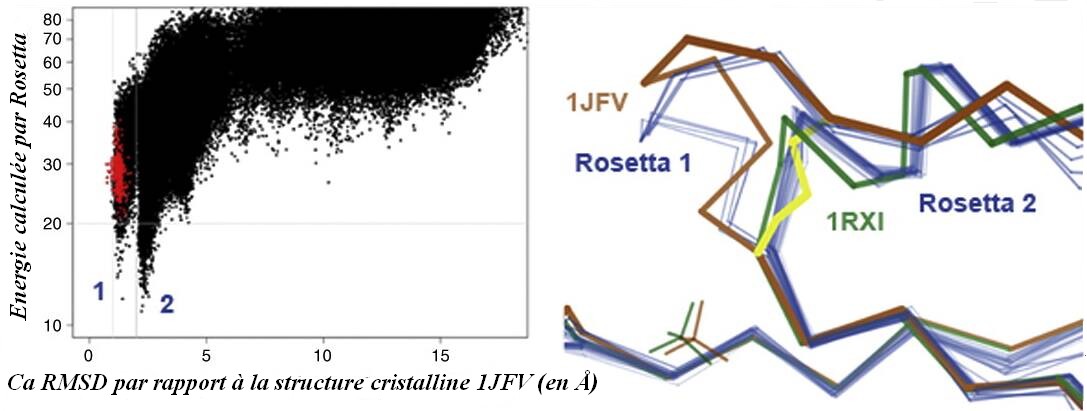
Découverte d'un état fonctionnel pertinent. Premier graphique : Score : CαRMSD pour une simulation de monomères isolés d'une arenate reductase (code PDB : 1JFV). L'axe des ordonnées donne l'énergie de repliement calculé par Rosetta, et l'axe des abscisses donne le CαRMSD de la structure cristalline. Les points rouges sont des modèles élastiques de la structure cristalline. Rosetta identifie 2 pics de basse énergie distincts, ce qui suggère la présence de 2 états isoénergétiques proches mais distincts dans la protéine réelle. Deuxième graphique : L'Arsenate reductase subit un pont disulfure Cys10–Cys82–Cys89 dans le cadre de sa réaction. La structure cristalline de l'oxyde 1JFV (brun) a une mutation C10S pour capturer l'extrêmité du pont. SS 82-89 (en jaune). La suite ici : Tableaux et graphiques (Figure 10) |
Cliquez ici pour accéder au résumé original de l'article en anglais. Vous pouvez accéder aux tableaux et graphiques de l'article en cliquant sur l'onglet "Figures/Tables"
Publication reçue le 2 Septembre 2010, révisée le 29 Octobre 2010, acceptée le 2 Novembre 2010. Disponible en ligne le 10 novembre 2010
Michael D. Tyka 1 , Daniel A. Keedy 2 , Ingemar André 3 , Frank DiMaio 1 , Yifan Song 1 , David C. Richardson 2 , Jane S. Richardson 2 et David Baker 1
1 Département de Biochimie, Université de Washington, Seattle, État de Washington 98195, Etats-Unis
2 Département de Biochimie, Université Duke, Durham, Caroline du Nord 27710, Etats-Unis
3 Université de Lund, Biochimie Structurale, Getingev. 60, S-221 00 Lund, Suède
Sommaire de l'article :
- Introduction
- Résultats
- Discussion
- Méthode
- Remerciements
- Références