


 Article en anglais (format .PDF)
Article en anglais (format .PDF)
Accès à l'article scientifique (en anglais, pdf de 92 Ko)
Nous venons de publier l'analyse initiale du très grand nombre de modélisations moléculaires issues de nos calculs sur FightAIDS@Home.
Cette semaine le premier article scientifique approuvé par comité de lecture a été publié, il rend compte des résultats de notre recherche FightAIDS@Home. Comme nous continuons à tester expérimentalement la première série de composés identifiés dans la première expérience informatique, nous avons pu analyser un plus large éventail de résultats informatiques pour fournir plus d'informations sur la diversité des mutations de la protéase virale du VIH et sur leur capacité de réponse à une large catégorie de composés chimiques divers.
Cette analyse, et la façon dont nous pouvons l'utiliser est le sujet de l'article qui parait dans le Journal of Chemical Information and Modeling. L'article est intitulé : "Analyse au moyen de l'outil informatique des liaisons entre les structures mutantes et de type sauvage du VIH et des ligands issus de diverses bibliothèques" Les auteurs sont Max Chang, Lindy Lindstrom, Arthur Olson, et Richard Belew.
Voici le résumé de cet article :
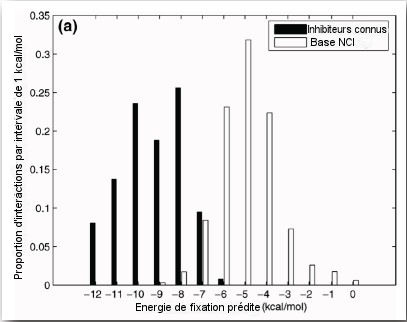
Le projet d'informatique répartie FightAIDS@Home utilise AutoDock comme un filtre initial des structures de la protéase du VIH en les comparant à 1771 ligands différents dont des inhibiteurs connus de la protéase et d'autres ligands issus de diverses bibliothèques. La masse des résultats permet de nouvelles analyses à grande échelle du profil de liaison énergétique des structures du VIH. Après avoir identifié les composés principaux, ces caractérisations fournissent des méthodes pour choisir les structures représentatives des protéines mutantes et de type sauvage dans un ensemble plus grand. A partir des profils d'énergie de liaison des structures issues de la banque de données des protéines (PDB : Protein Data Bank), une analyse des composants principaux (PCA) a identifié 7 collections de protéases. L'analyse complémentaire a constaté que la structure de la protéase de type sauvage 2BPZ capture le mieux la tendance centrale de l'ensemble des protéases. Comparer des inhibiteurs de protéase connus par rapport à un ensemble de ligands différents place le seuil significatif de l'énergie de liaison AutoDock à -7.0 kcal/molécule pour distinguer les ligands de liaison fortement significatifs et d'autres liaisons à énergie faible/non caractéristique. Ce seuil retient presque 98% des interactions inhibitrices connues tout en rejetant plus de 95% des interactions suspectées d'être non inhibitrices. Ces méthodes devraient en général être utiles dans des projets virtuels de criblage et pourront être utilisées pour améliorer les prochaines expériences du projet FightAIDS@Home.
En d'autres termes, les résultats de FightAIDS@Home nous ont donné la possibilité de classer à la fois les protéases mutantes ainsi que des inhibiteurs chimiques potentiels. De cette façon les calculs que nous réaliserons dans le futur seront plus efficaces et plus ciblés. Nous serons par exemple capables d'utiliser un petit ensemble de protéases mutantes pour représenter un ensemble plus large de VIH mutants dans nos modélisations moléculaires. Ceci nous permettra d'examiner un plus large éventail de composants chimiques et ceci beaucoup plus efficacement, ou d'utiliser des modèles de structures mutantes plus sophistiqués, qui intègreront de la flexibilité dans leur représentation.
En fait il s'agit là d'une avancée technique pionnière, cela signifie que nous allons obtenir des résultats plus fiables et plus significatifs dans les calculs que nous allons maintenant effectuer. Ceci devrait nous rapprocher de notre but qui est de trouver de nouveaux agents thérapeutiques anti-VIH qui soient efficaces face à la résistance aux médicaments. Ces résultats n'auraient pas été possibles sans la puissance de calcul informatique du World Community Grid, et le généreux soutien des volontaires au projet FightAIDS@Home. Nous sommes très reconnaissants de vos efforts, et votre contribution est reconnue dans l'article scientifique.
|
|
Distribution des énergies de liaison pour les inhibiteurs et les composés issus de la base de données du NCI (National Cancer Institute)
|
Prof. Arthur J. Olson
Dr. Garrett M. Morris
Dr. William Lindstrom
Alexandre Gillet
Dr. Richard K. Belew
Max W. Chang
Références :
Max W. Chang, William Lindstrom, Arthur J. Olson, et Richard K. Belew (2007) “Analysis of HIV Wild-Type and Mutant Structures via in Silico Docking against Diverse Ligand Libraries” J.Chem. Inf. Model.