



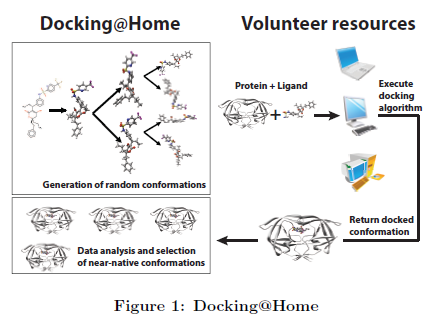
2- Explorer le vaste espace de conformations des molécules ligand avec Docking@Home
(D@H) est un projet de calcul bénévole visant à construire par le calcul distribué un environnement propice afin d’aider les scientifiques à mieux comprendre les interactions protéine-ligand. En choisissant avec précision les détails atomiques ainsi que les structures de ligand. D@H utilise des milliers d’ordinateurs bénévoles pour simuler le comportement de ces petites molécules (appelées ligand) lors de leur amarrage au sein d’une protéine et ainsi contrôler leurs fonctions. D@H s'appuie sur BOINC [1] (Berkeley Middleware Open Infrastructure for Network Computing) qui génère, distribue et réceptionne les données par internet. Au sujet plus particulier de Docking@Home, celui-ci distribue les travaux contenant un ligand et une protéine aux machines bénévoles (également appelées clients D@H). La simulation d'amarrage est effectuée sur l’ordinateur-hôte, qui, à la fin du calcul, retourne la conformation du ligand, lorsque celle-ci s'est amarrée dans la protéine. Par conformation de ligand, nous entendons la position tridimensionnelle des atomes de ligand et leurs interactions avec la protéine. Actuellement, D@H est pris en charge par environ 12'000 à 30'000 bénévoles et leurs ordinateurs; D@H a récupéré plus de 2TBytes de données en six mois et a recueillis quotidiennement environ 30’000 résultats d'amarrage. Ceux-ci sont stockés dans un référentiel et analysés par la suite, ainsi nous sélectionnons parmi des millions de candidats un ensemble très réduit de conformations du ligand en se basant sur la probabilité de la convergence de l'algorithme d'amarrage.
Le solvant, usuellement de l'eau, dans lequel les complexes protéine-ligand résident, a une influence fondamentale sur les interactions de celles-ci dans toute simulation d'amarrage. Le solvant agit sur les interactions électrostatiques de dépistage et au sein des atomes dans la structure moléculaire. D@H utilise deux algorithmes d'amarrage avec différentes formes du solvant:
• Méthode 1: Une représentation implicite de l'eau est utilisée avec un coefficient diélectrique dépendant de la distance (faible si la distance entre les atomes est étroite et progressivement plus grande si la distance est croissante)
• Méthode 2: Une représentation implicite plus physiquement précise de l'eau est utilisée à l'aide d'un modèle quasi-authentique.
La méthode basée sur le modèle quasi-authentique est plus intensive pour le CPU et la mémoire. En même temps elle fournit une description physiquement plus précise du potentiel de l'énergie d'un ligand à l’endroit précis de la conformation de celle-ci, lorsqu’elle est exposée au solvant. Dans de nombreuses situations, où une grande partie du ligand est exposé au solvant, le modèle authentique devrait contribuer significativement à fournir la meilleure conformation du ligand (par exemple lorsqu'une orientation d'un ligand donnée laisse un groupe volumineux hydrophobe exposé au solvant, cela la pénalise, exposer un groupe hydrophile comme un groupe hydroxyle OH au solvant est beaucoup plus favorable). Les travaux récents (de Rahaman, Armen, Estrada, Taufer et Brooks) afin de comparer l'exactitude de la méthode 1 et la fabrication des molécules de la méthode 2 ont démontré que la mise en œuvre particulière utilisée dans ce travail a des performances plus pauvres pour distinguer les modèles quasi-authentiques au niveau géométrique. Cette observation est similaire à celle observée par les calculs effectués sous Boinc. Les créations de molécules quasi-authentiques de la méthode généralisée sont capables d’être plus performantes que la méthode 1. Toutefois, dans le scénario actuel, même compte tenu de la précision moindre de la méthode 2, nous démontrons que notre regroupement hiérarchique probabiliste est en mesure de façon significative d’améliorer la discrimination des conformations quasi-native, même compte tenu de l'incertitude inhérente de la fonction pour la méthode 2.
D@H cible trois protéines différentes : la trypsine, le VIH et p38 Alpha. Ces protéines ont été sélectionnées parce qu’elles se caractérisent par différents degrés de flexibilité au cours du processus d'amarrage. La trypsine [7] est une protéase relativement rigide qui brise les autres protéines dans le système digestif. Des études récentes suggèrent que les inhibiteurs de la trypsine peuvent avoir des applications potentielles dans le traitement du cancer du sein. Il a été observé que la trypsine active les récepteurs des protéases (protéine dans la membrane de la cellule tumorale). Lors de ce processus, la protéine provoque la dégradation de la matrice extracellulaire, résultant de la propagation de la cellule tumorale d’un endroit à l'autre (métastases). Des médicaments peuvent agir comme inhibiteur en désactivant la trypsine protéase et sont donc des agents potentiels capables d'arrêter la propagation du cancer du sein. La protéase du VIH (VIH PR) [2] est une protéine relativement flexible au sein de la molécule du VIH et est essentielle à sa réplication dans les cellules humaines. Au cours du processus de construction du virus VIH à l'intérieur de la cellule humaine, le VIH PR Clive, nouvellement synthétisé pourrait s’accrocher au sein de la molécule en ciblant le génome infecté. Ce processus est nécessaire pour construire un virus VIH mature. Le VIH PR est une cible thérapeutique- bien connue pour le traitement de l'infection au VIH et afin de prévenir l'apparition du sida chez les patients déjà infectés. Un médicament qui peut se lier étroitement à la molécule active inhibera significativement l'activité enzymatique d’une importante population de molécules de VIH PR et réduire massivement le processus de réplication virale dans les cellules infectées. Ces médicaments sont appelés des inhibiteurs de protéase. Plusieurs inhibiteurs de protéase, comme saquinavir, ritonavir, indinavir, et Nelfinavir sont disponibles pour le traitement de l'infection par le VIH.