



5- Précision des mesures de DOCKING
Pour tester notre cadre probabiliste hiérarchique, nous avons réalisé des essais « d’accueil » pour chacun des 23 complexes protéine-ligand de la protéase du VIH (c.-à-1ajv, 1ajx, 1d4h, 1d4i, 1d4j, 1ebw, 1ebz, 1g2k, 1g35, 1gno, 1hbv, 1hih, 1hps, 1hpx, 1hsg, 1htf, 1hvi, 1hvj, 1hvk, 1iiq, 1m0b, 1ohr, et 1T7K), 21 ligands d'accueil dans la protéine trypsine (c.-à-1c2d, 1c5p, 1c5q, 1ce5, 1g36, 1GHz, 1gi4, 1gi6, 1gj6, 1k1i, 1k1j, 1k1l, 1k1m, 1k1n, 1ppc, 1pph, 1qb6, 1tpp, 1xug, 2bza, 3ptb), et 12 ligands d'accueil dans la kinase P38alpha (c.-à-1a9u, 1bl6, 1bl7, 1di9, 1kv1, 1kv2, 1ouk, 1ouy, 1oz1, 1w83, 1w84, 1yqj). Pour chacun de ces complexes nous avons réalisé 2 millions d’essais.
Dans une première série de tests afin d'évaluer si nos groupes probabilistes hiérarchiques tiennent la route et peuvent capturer des conformations quasi-native indépendamment de la méthode d'accostage, nous avons considéré les deux méthodes d'accueil décrites à la section 3 (méthode 1 et 2) et généré de façon aléatoire des ligands comme des conformations initiales (voir les figures 5 et 6). Dans une deuxième série de tests, on a évalué si les configurations initiales utilisées dans le procédé d'accueil jouent un rôle actif dans la polarisation de l'exactitude de notre sélection de groupe de base, nous avons utilisé la méthode 1 et les ligands définis par l'utilisateur dont les conformations sont > 5 Angstroms à partir de la structure cristalline correct (voir figure 7). Notez que la conformation avec > 5 Angstroms des structures cristallines correspondantes est considérée comme une mauvaise conformation.
Nous avons utilisé notre groupe probabiliste hiérarchique pour trouver les conformations de ligands les plus susceptibles d’être quasi-native pour chaque complexe. Pour chaque complexe, notre cadre d’étude c’est réalisé sur 100.000 conformations de ligands. La distance métrique utilisée pour grouper chaque ligand est le RMSD de ses coordonnées atomiques contre tous les autres ligands déjà dans le groupe. Si une simulation converge, alors le plus grand groupe avec une variance interne inférieure (notée comme un groupe cible) est probablement le groupe qui contient le plus de conformations quasi-native. Dans nos expériences, la conformation ligand avec le plus haut degré d'appartenance (centroïde) pour le groupe cible est sélectionnée comme notre prédiction quasi-native (voir équation 1). Dans le reste de cet article, nous nous référons à cette conformation comme le regroupement candidat d’un complexe donné.
L'ensemble du processus de regroupement et de sélection du groupe candidat a été réalisé sans l'aide des structures cristallines disponibles pour les complexes de BDPT [15]. Les structures cristallines ne jouent un rôle important que dans la phase de validation de notre cadre quand, pour chaque complexe, nous avons calculé le RMSD du groupe candidat à l'égard de sa structure cristalline.
Une conformation peut être considérée comme une conformation quasi-native si son RMSD est inférieure ou égale à deux Angstroms, mais des conformations RMSD entre deux et trois Angstroms sont considérées des résultats intéressants. Dans le cas de l'approche énergétique, nous considérons que nous capturons une conformation quasi-native si la médiane arithmétique est inférieure ou égale à deux Angstroms. L'utilisation de la médiane est préférable que la moyenne, car moins affectée par les valeurs extrêmes [9].
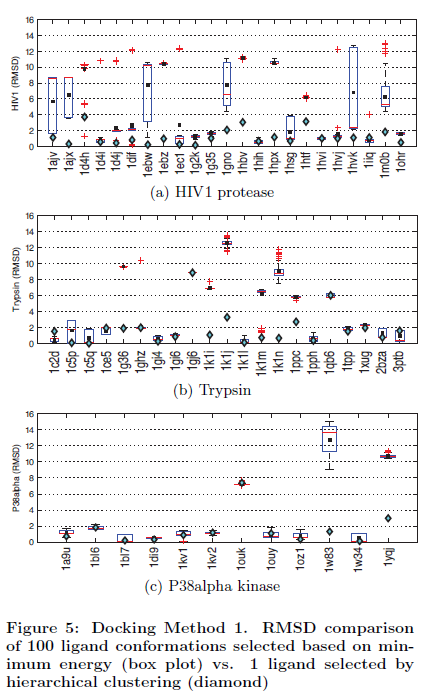
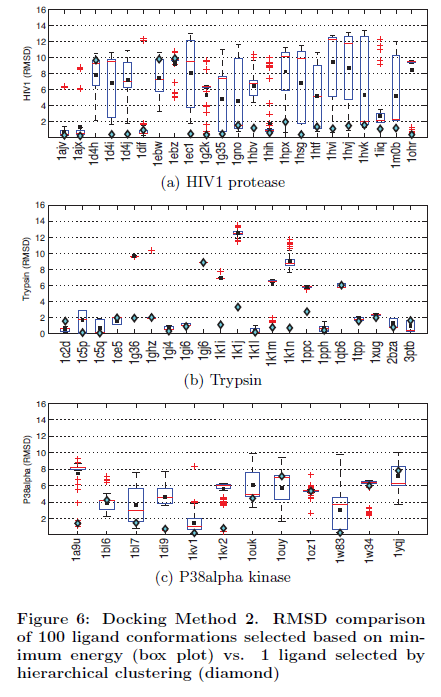
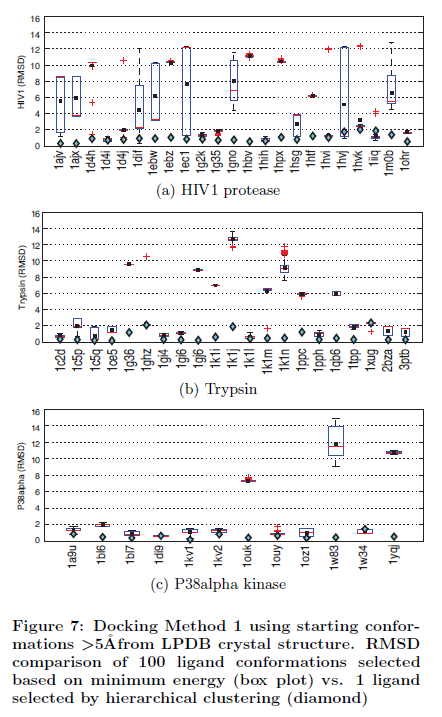
Les figures 5.a, b et c présentent les deux comparaisons de validation pour l’intérêt des trois protéines pour la méthode 1. Les figures 6.a, b et c présente les deux comparaisons pour les mêmes trois protéines avec la méthode 2, et les figures 7.a, b et c présentent les comparaisons pour les trois mêmes protéines avec la méthode 1 avec une conformation définie par l'utilisateur qui a été de moins de 5 Angstroms d’espacement de la structure cristalline. Les figures 5.a, 6.a et 7.a se rapportent à la protéase du VIH1, les figures 5.b, 6.b et 7.b se référent à la trypsine, et les figures 5.c, 6.c et 7.c se rapportent à P38alpha kinase. Sur l’axe des abscisses, nous montrons les différents complexes et sur l’axe des ordonnées, nous montrons leur RMSD, le plus faible étant le meilleur. Les losanges représentent les RMSD du groupe candidat WRT de la structure cristalline. Les graphiques représentent la boîte du RMSD des 100 conformations choisis en fonction de l'énergie.
La boîte de données graphiques se compose de sept différents éléments d'information. Les favoris du bas s’étendent du 10e pourcentage (décile inférieur) au 90e pourcentage supérieur (décile supérieur). Les valeurs aberrantes sont placées à la fin des déciles supérieurs favoris. Le haut, le bas, et le trait au milieu de la boîte correspond au 75e pourcentage (en haut), 25e pourcentage (en bas), et 50e pourcentage (au milieu). Un carré indique la moyenne arithmétique.
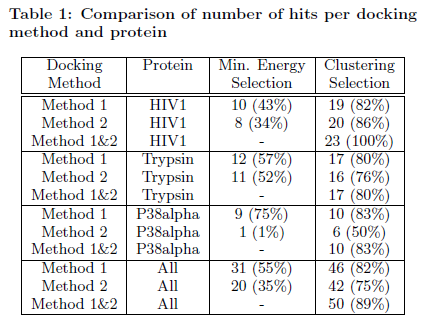
Pour la méthode 1 avec la protéase du VIH1, l'approche énergétique est capable d'identifier seulement 10 des 23 conformations quasi-native (soit un taux de succès de 43%) tandis que notre méthode de clustering capte 19 des 23 conformations quasi-native (soit un taux de succès de 82%). Pour la trypsine, l'approche énergétique identifie seulement 12 des 21 conformations quasi-native (taux de succès de 57%) et notre méthode de clustering capte 17 des 21 conformations quasi-native (taux de succès de 80%). Pour la kinase P38alpha, l'approche énergétique identifie 9 des 12 conformations quasi-native (taux de succès de 75%) et notre méthode de clustering remporte 10 des 12 conformations quasi-native (taux de succès de 83%). Lorsque nous considérons la méthode 2, la plus intensive en calculs, pour la protéase du VIH, l'approche énergétique est capable d'identifier seulement 8 des 23 conformations quasi-native (taux de succès de 34%) alors que notre méthode de clustering capte 20 des 23 quasi-native conformations (taux de succès de 86%). Pour la trypsine, l'approche énergétique identifie seulement 11 des 21 conformations quasi-native (taux de succès de 52%) et notre méthode de classification représente 16 des 21 conformations quasi-native (taux de succès de 76%). Pour la kinase P38alpha l'approche énergétique identifie 1 des 12 conformations quasi-native (taux de réussite de 0,8%) et notre approche prend en compte 6 des 12 conformations quasi-native (taux de succès de 50%). Le tableau 1 récapitule les taux de succès pour les deux méthodes d'amarrage. Comme le montre le tableau, dans le cadre de l’étude, notre approche surpasse l'approche énergétique pour tous les complexes et pour chaque méthode. Grâce à notre méthode de classification, nous pouvons voir qu'aucune des deux méthodes d'accueil sont nettement supérieures à l'autre. La combinaison des deux méthodes d'amarrage (méthode 1 et 2) permet de renforcer encore la précision de nos prédictions pour la protéase du VIH pour lesquels nous avons observé un taux de succès de 100%. Pour les deux autres protéines, nous avons observé le même taux de succès. Dans le tableau, nous ne comparons pas la sélection de l'énergie en fonction basée en combinant les deux méthodes d'accueil, car elles sont composées d'approximations tout à fait différentes de l'énergie potentielle.
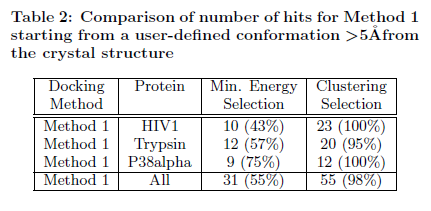
Le tableau 2 résume les taux de succès pour la Méthode d’accueil 1 lorsque le processus d'accueil commence à partir d'une conformation définie par l'utilisateur à partir d'au moins 5 Angströms à partir de la conformation de la structure cristalline BDPT. Pour la protéase du VIH1, l'approche énergétique est en mesure d'identifier que 10 des 23 conformations quasi-native (taux de succès de 43%) tandis que notre méthode de classification englobe les 23 conformations quasi-native (taux de succès de 100%). Pour la protéine trypsine, l'approche énergétique est en mesure d'identifier que 12 des 21 conformations quasi-native (taux de succès de 57%) tandis que notre méthode de clustering capte 20 des 21 conformations quasi-native (taux de succès de 95%). Pour la kinase P38alpha, l'approche énergétique identifie 9 des 12 conformations quasi-native (taux de succès de 75%) et notre approche prend en compte 12 des 12 conformations quasi-native (taux de succès de 100%). Lorsque l'on considère les trois ensembles de complexes avec l'approche énergétique nous avons un taux de succès de seulement 55% alors que notre méthode de regroupement identifiés à proximité de la conformation native-ligand est bonne dans 98% des cas. Comme le montre le tableau 2, notre cadre surpasse toujours l'approche basée sur l'énergie pour les deux ensembles de complexes et montre des tendances similaires comme pour la méthode 1 quand nous utilisons des conformations de ligand de départ aléatoires qui sont générées par MD à partir de la conformation ligand cristallographique. En outre, les données du tableau 2 montrent que les conclusions finales de cette étude sont robustes et non influencées par le fait de commencer la recherche de conformation par la conformation quasi-native du ligand initial.
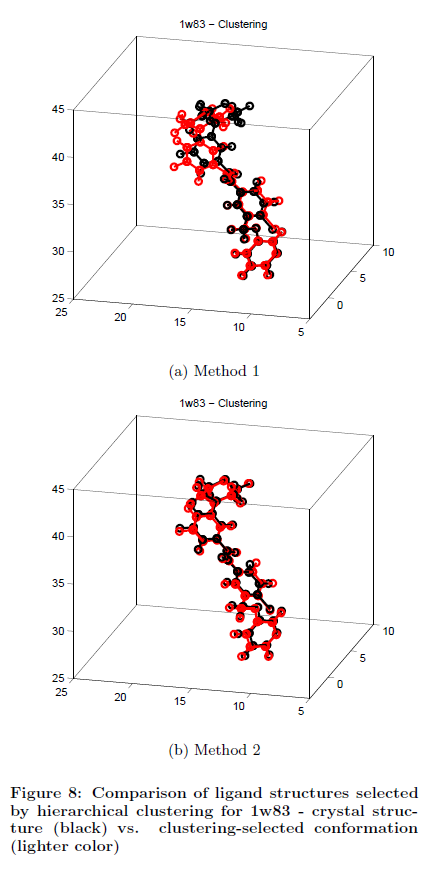
Si l'on reprend le même complexe (1w83) présenté dans la figure 3, et que nous utilisons cette fois notre méthode de classification pour la sélection de la conformation candidate, nous constatons que nous sommes en mesure de trouver une conformation quasi-native pour les deux méthodes d'amarrage (Figure 8.a pour la méthode 1 et la figure 8.b pour la méthode 2). Le ligand noir, représentant le ligand dans la structure cristalline BDPT, chevauche pratiquement le ligand gris, qui représente dans ce cas la conformation ligand candidate sélectionnés par notre classification hiérarchique probabiliste. Contrairement à la sélection ligand fondées sur l'énergie, notre regroupement probabiliste est capable d'identifier avec précision la conformation ligand quasi native indépendamment de la méthode d'accostage.
Pour mieux illustrer le comportement de notre méthode de clustering et de sa capacité à identifier un nombre variable de clusters dynamique, la figure 9 montre le résultat pour neuf des complexes en utilisant la méthode 2 (trois pour le VIH,, 1d4i, 1dif, et 1ebw; trois pour la trypsine, -1k1m, 1c2d, et 3ptb, et trois pour P38alpha,, 1a9u, 1oz1, et 1ouy). Pour chaque protéine, nous présentons un complexe pour laquelle notre méthode surpasse clairement l'approche naïve (colonne de gauche), un complexe pour laquelle notre méthode a une précision similaire à l'approche naïve (colonne centrale), et un complexe pour lequel l'approche naïve a une meilleure précision (colonne de droite). Nous utilisons la méthode 2, puisque la méthode 1 est toujours dans notre cadre soit supérieur ou égal à la précision de l'approche naïve (voir Figure 5). Ce n'est pas toujours le cas pour la méthode 2 (voir Figure 6). Après le regroupement terminé, nous avons la cartographions chaque conformation de chaque groupe par rapport à son énergie (abscisse) et ses RMSD par rapport à la structure cristalline (ordonnée). Les différentes couleurs indiquent les différents groupes (comme l'indique la légende). Le groupe le plus profond (zone de gris) dans la hiérarchie est le groupe cible contenant le candidat montré comme une ligne solide horizontale. La meilleure conformation choisis en fonction de son énergie minimale est indiquée par une ligne horizontale en pointillés. Comme le montre la figure 9, la profondeur du cluster hiérarchique (nombre de grappes) est variable, allant de deux pour, par exemple, 1dif, à quatre, par exemple pour, 1d4i, et dépend du paysage énergétique du complexe [6]. Le nombre maximal de clusters dans l’ensemble de données D @ H est de six. Aucune intervention humaine n'est nécessaire pour définir la profondeur de clustering.

En raison de son taux de réussite élevé, le cadre proposé peut être utilisée sans être changé pour sélectionner des conformations de ligand quasi-native dans les simulations d'amarrage protéine-ligand lorsque les structures cristallines sont inconnus. En outre, le cadre peut être utilisé pour la sélection de conformations quasi-native dans d'autres simulations pertinentes, telles que le repliement des protéines et la prédiction des protéines.