



3- Sélection de conformations quasi-native sous incertitude
L’amarrage protéine-ligand utilise des méthodes d’évaluation distinctes pour deux tâches : la première étape est la discrimination de la géométrie de liaison du ligand (l'identification de conformations quasi-natales) et la deuxième étape est une comparaison de ligands différents (d’espèce chimique différente) pour prévoir quel ligand se lie le mieux à la protéine. D@H est un moteur pour la première étape et les résultats pour la deuxième étape peuvent être réalisés dans une étape post-traitement. Cet article ne se concentre pas sur la deuxième étape, mais est entièrement concentré sur la première.
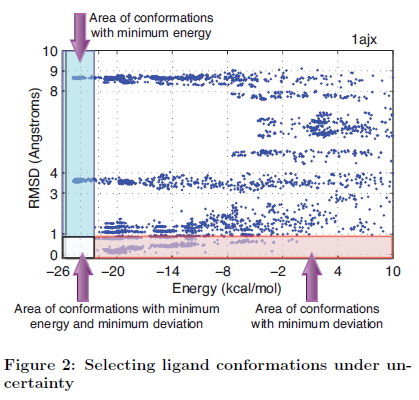
Tout en s’occupant de la méthode de notation, nous avons initialement compté sur une approche d’évaluation traditionnelle basée sur des valeurs d'énergie : nous avons choisi des ligands avec une énergie faible comme les conformations quasi-natales les plus probables. Nous avons immédiatement identifié les insuffisances de cette approche en termes d'exactitude. La figure 2 montre un exemple de 100,000 conformations ligand (chaque point dans la figure est une conformation ligand) obtenu avec Docking@Home pour le complexe 1ajx. Ici, les conformations ligand sont marquées par leur énergie potentielle (abscisse) et leur "Racine Carrée Moyenne Ecart-type" (RMSD) en ce qui concerne la structure cristalline connue (l'axe des ordonnées). Le RMSD est mesuré en Angströms (°A) et est calculé par la racine carré de la moyenne de la différence élevée au carré de tout atomes de ligand non-hydrogène dans la simulation de conformation ligand et les atomes de ligand dans la structure cristalline. La figure montre trois régions de pertinence : (1) Le secteur de conformations avec l'énergie minimale, qui est le rectangle vertical qui va de-26 à -22 kcal/mol. Une conformation ligand avec l'énergie minimale ne fait pas toujours une conformation quasi-natale. Les conformations dans ce secteur seraient choisies par une méthode qui représente seulement l'énergie et il y aurait des chances que ces candidats ne soient pas des conformations quasi-natales. Dans la figure nous pouvons voir deux secteurs de minimums entre 3-4°A et 8-9°A. (2) Le secteur de conformations avec un écart minimal (RMSD). Le RMSD est calculée par rapport à la structure cristalline, comme expliqué ci-dessus. Ce secteur est noté par le rectangle horizontal qui va de 0 à 1 °A. Idéalement, l’évaluation minimum d'une fonction avec une importante exactitude se trouverait dans ce secteur. Cependant, le minimum global n'est pas toujours trouvé (c'est le cas dans la Figure 3). Pour la découverte de nouveaux médicaments, la dimension d'écart (l'axe des ordonnées) est inconnue et ne peut pas être utilisée pour choisir le candidat de conformation de ligand. Dans cet article nous supposons que cette restriction se tient toujours et nous utilisons le RMSD seulement pour des buts de validation. (3) est le secteur de conformations avec l'énergie et l'écart minimal, qui est l'intersection des deux autres secteurs décrits auparavant. Idéalement ce secteur devrait être très peuplé pour augmenter les chances de choisir le bon candidat de conformation de ligand. Comme le montre la figure, ce n'est pas le cas, l'augmentation du niveau d'incertitude permet de rendre plus difficile la sélection des candidats ligand quasi-native.
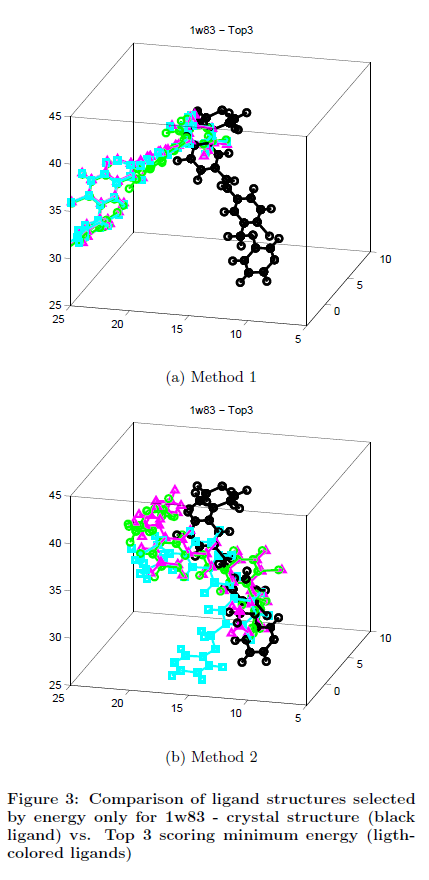
Nous avons observé le problème à travers les résultats d’amarrage générés par les deux différentes méthodes pour les trois protéines et les différents ligands considérés. La figure 3 montre un exemple de ce phénomène. 1w83 est la p38alpha kinase dans un complexe avec une petite molécule inhibitrice (ligand). Les deux sous-figures, 3.a et 3.b, montrent la comparaison graphique de la 1w83 seul ligand dans la structure cristalline (ligand noir) par rapport au top 3 des conformations ligand marqué par le minimum d'énergie sur l'ensemble des échantillons D H @ pour ce complexe (ligands gris). La figure 3.a correspond à la conformation ligand produit par la méthode 1. Il s'agit d'un cas extrême où la fonction de notation attribue le plus bas de l'énergie à une série de conformations qui convergent avec une orientation nettement différente de la structure cristalline. Figure 3.b correspond à la conformation ligand produit par la méthode 2 pour le même complexe. Cette figure montre que le minimum d'énergie des structures ne convergent pas vers une solution unique en dépit du grand nombre d'échantillons D H @. Dans le même temps, ces trois résultats sont sensiblement différents entre eux et aucun d'eux n'est suffisamment précis pour être appelé une conformation quasi-native.
Nous excluons que le problème d’incertitude de notation est liée à un échantillonnage insuffisant ou inefficace de l'espace de travail: D @ H est en effet capable d'échantillonnage intensif de l'espace d’étude. D'autre part, la modélisation des énergies est encore imprécise, même en utilisant la méthode basée sur le Generalized Born modèle du solvant implicite.